
One of the questions I most often receive: Does being in ketosis automatically translate to fat loss?
For those too busy to read ahead, let me give you the punch line: No. For those who want to understand why, keep reading (hopefully this is still everyone). This topic is — surprise, surprise — very nuanced, and almost always bastardized when oversimplified, which I’m about to do, though hopefully less than most. Without oversimplifying, though, this will turn into a textbook of 1,000 pages.
From the ketosis series, or at least the first and second part, along with the video in this previous post, you should have taken away that ketosis is not some ‘magical state of mystery.’ It’s simply a state of physiology where our liver turns fatty acid (both ingested and stored) into ketones.
There seems to be great confusion around ‘nutritional’ ketosis (a term we use to distinguish ‘dietary-induced’ ketosis from the other 2 forms of ketosis: starvation ketosis and ketoacidosis, the latter a serious complication of type I diabetes). But, before I try to dispel any of the confusion, we need to go through a little primer on what I like to call “fat flux.”
One point before diving in, please do not assume because I’m writing this post that I think adiposity (the technical term for relative amount of fat in the body) is the most important thing to worry about. On the contrary, I think the metabolic state of the cell is far more important. While there is a correlation between high adiposity (excessive fat) and metabolic dysfunction, that correlation is far from perfect, and, as I’ve discussed elsewhere, I think the arrow of causation goes from metabolic dysfunction to adiposity, not the reverse. But, everyone wants to lose fat, it seems, so let’s at least get the facts straight.
Let’s start with an assertion: Barring the presence of scientific evidence I’m unaware of, and barring surgical intervention (e.g., liposuction), reducing the adiposity of a person is achieved by reducing the adiposity of individual adipose cells, collectively. In other words, the number of adipocytes (fat cells) we have as an adult does not change nearly as much as their size and fat content. So, for people to reduce their fat mass, their fat cells must collectively lose fat mass.
Fat flux 101
According to “An Etymological Dictionary of Modern English,” the word flux comes from the Latin word fluxus and fluere, which mean “flow” and “to flow,” respectively. While the term has a clear mathematical meaning in physics, defined by a dot product I promise I won’t speak of, you can think of flux as the net throughput which takes into account positive and negative accumulation.
If we start with a bucket of water and put a hole in the bottom, the result, needless to say, is an efflux of water, or negative water flux. Conversely, if we start with a bucket – no hole – and we pour water in, that’s an influx of water, or positive water flux.
If that makes sense, then the idea of fat flux is pretty straight forward. If more fat enters a fat cell (called an adipocyte) than leaves it, the fat cell is experiencing a net influx – i.e., positive fat flux. And, if more fat leaves a fat cell than enters, the reverse is true: it is experiencing a net efflux, or negative fat flux.
Not surprisingly, a fat cell is more complicated than a bucket. Basically, though, there are two “inputs” and one “output.” The figure below shows this in some detail. (TAG stands for triacylglycerol, which is another word for triglyceride, which is the storage form of fat.) The first thing you may appreciate, especially since I’ve highlighted it, is the role insulin plays in regulating the process of fat flux. Insulin does the following:
- Upregulates lipoprotein lipase (LPL), an enzyme that breaks down TAG so they can be transported across cell membranes. Since TAG are too big to bring across cell membranes, they need to be “hydrolyzed” first into free fatty acids, then re-assembled (re-esterified) back into TAG.
- Translocates GLUT4 transporters to the plasma membrane from endosomes within the cell. In other words, insulin moves the GLUT4 transporter to the cell surface to bring glucose into the cell.
- Facilitates lipogenesis, that is, facilitates the conversion of glucose into acetyl CoA which gets assembled into fatty acids along with glycerol.
- Facilitates esterification, that is, facilitates the process of assembling fatty acids into TAG (3 fatty acids per TAG).
- Inhibits hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL), two important enzymes that breaks down TAG into fatty acids and glycerol such that the fatty acids can be released from the fat cell. Once bound to albumin the free fatty acids are free to travel elsewhere in the body for use (e.g., to the liver for conversion to ketones, to the heart muscle or skeletal muscles for conversion to ATP).
- Though not shown in this figure, insulin appears to indirectly act on malonyl-CoA, a potent inhibitor of CPT I, one of the most important mitochondrial enzymes that facilitates the oxidation of fatty acids. (CPT I is what enables fatty acids to be shuttled into the mitochondria for oxidation, the process which releases or liberates their energy through electron transport.)
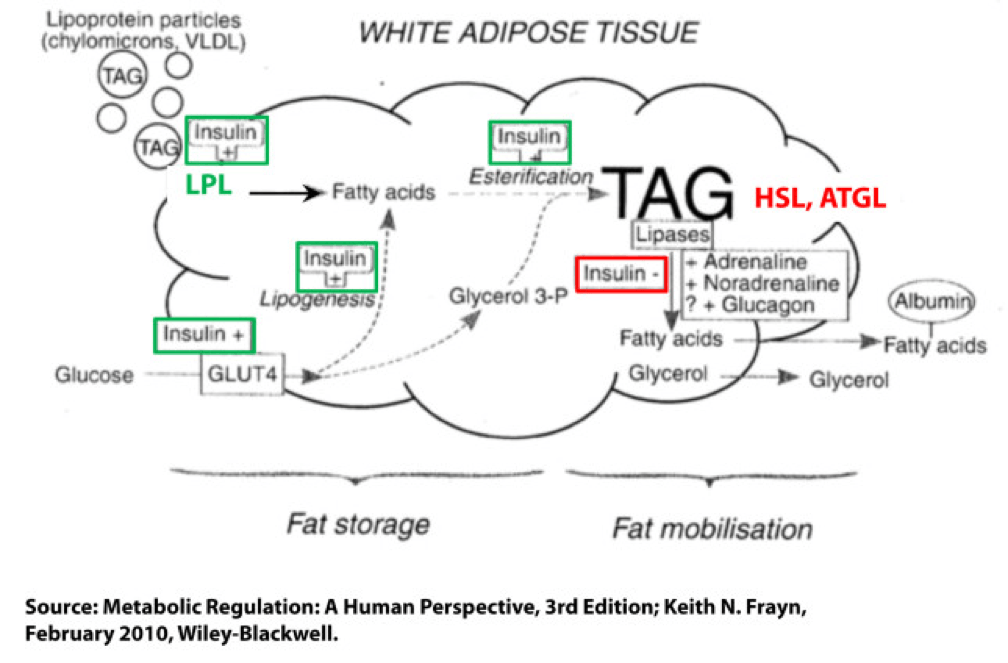
Other hormones and enzymes in the body also play a role. For example, under a sympathetic response, the so-called “fight or flight” response, adrenaline and noradrenaline (i.e., epinephrine and norepinephrine) activate HSL and ATGL to combat the effect of insulin as an inhibitor of lipolysis, thereby increasing lipolysis, or liberating stored energy from the fat cell. Glucagon may also play a role in this process, though the exact role is not as well understood, at least not in humans.
So in summary, insulin is indeed the master hormone that regulates the flow of fat (and glucose) into and out of a fat cell. There are other players in this game, to be sure, but insulin is The General. High levels of insulin promote fat storage and inhibit fat oxidation, and low levels of insulin promote fat mobilization or release along with fat oxidation.
If this sounds crazy – the notion that insulin plays such a crucial role in fat tissue — consider the following two clinical extremes: type 1 diabetes (T1D) and insulinoma. In the former, the immune system destroys beta-cells (the pancreatic cells that make insulin) – this is an extreme case of low insulin. In the case of the latter, a tumor of the beta-cell leads to hypersecretion of insulin – this is an extreme case of high insulin. Prior to the discovery of insulin as the only treatment, patients who developed T1D would become emaciated, if the other complications of glycosuria and dehydration didn’t harm them first. They literally lost all fat and muscle. Conversely, patients with insulinoma often present looking not just obese, but almost disfigured in their adiposity. Because Johns Hopkins is a high-volume referral center for pancreatic surgery, it was not uncommon to see patients with insulinoma when I was there. As quickly as we would remove these tumors, the patients would begin to return to their previous state and the adipose tissue would melt away.
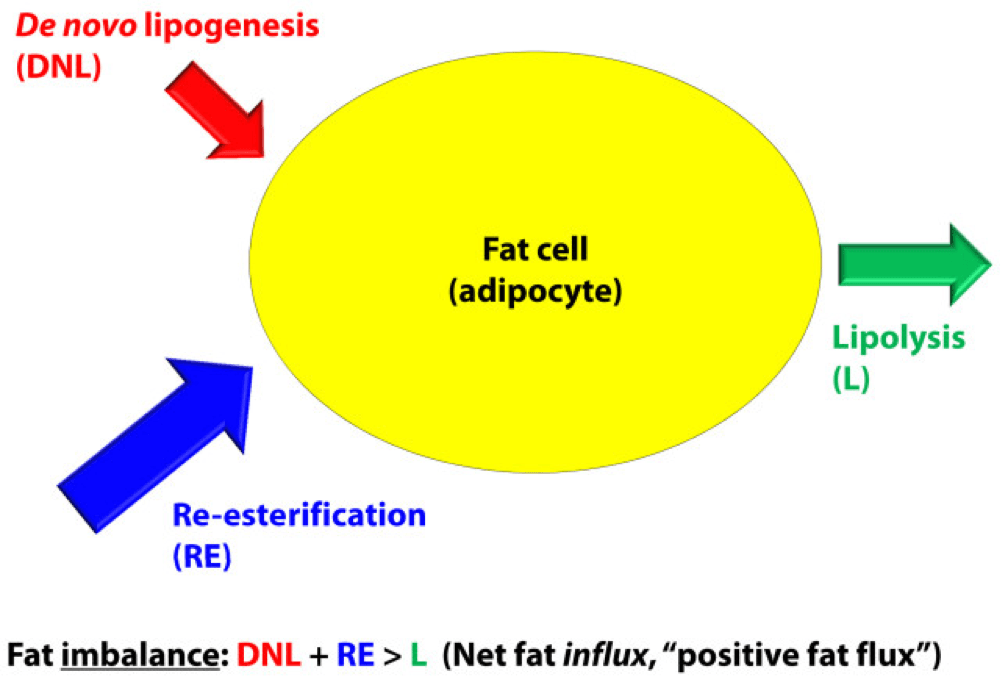
For the purpose of our discussion, I’ve simplified the more detailed figure above into this simplified figure, below. I’ve tried to size the arrows accordingly to match their relative contributions of each input and output.
The first figure, below, shows a state of fat balance, or zero net fat flux.

Input #1: De novo lipogenesis, or “DNL” – Until the early 1990’s there was no way to measure this directly, and so no one really had any idea how much this process (i.e., the conversion of glucose to fat) contributed to overall fat balance. Without going into great technical detail, , arguably one of the world’s foremost authorities on metabolomics and DNL, developed a tracer technique to directly measure this process. If I recall correctly, the original report was in 1991, but this paper is a great summary. Published in 1995 in the Journal of Clinical Investigation, this paper would go on to become the “citation classic.” This study demonstrated that under eucaloric feeding conditions, with about 50% of energy coming from CHO, DNL did not represent a significant contribution to fat flux. It was about 5%, hence the tiny red arrow under a state of fat balance (i.e., a state where fat entering the fat cell is equal to fat leaving the fat cell). A very important point to be mindful of, however, is this: this represents an average throughout the body and does not differentiate specifically between, say, DNL in the liver and DNL in the periphery (i.e., fat cells). This limitation is not trivial, but rather than focus on the very specific details of this paper, I’d rather use it as a framework for this discussion. (This paper is really interesting, and were it not for the fact that this post is going to be long enough, I would say much more about it. As such, I will probably do a full post on this paper and related topic in the future. The 1995 paper also examined what happened to DNL during periods of over- and under-feeding CHO and fat.)
Input #2: Re-esterification, or “RE” – In a state of fat balance, RE is largely composed of dietary fat sources that are not immediately used, but rather stored for later use. (Nuanced point: RE also includes fatty acids that were previously liberated from adipocytes, not oxidized, and are now being recycled back into TAG. This is a normal consequence of fat liberation. The fat cell probably ‘deliberately overdoes it’ by liberating more fatty acid from TAG just to be safe; that which is not oxidized is re-esterified. The exact balance of RE composed from dietary sources versus recycled fatty acids will depend on fat consumption and energy demands of the person. For the purpose of simplicity, this diagram does not show some portion of the L fraction returning to the RE fraction, though this is exactly what is happening in ‘real life.’)
Obviously, though, the relative size of the blue arrow depends on how much fat one is consuming and how many metabolic demands are in place for fatty acids. The latter is highly determined by dietary composition (see the discussion on RQ, or respiratory quotient, at about minute 31 in this video).
For the real aficionado, there is another wee bit of nuance here. This study, published in 1991 in the Journal of Lipid Research, suggested that the RE process is a bit more complicated than simply re-assembling fatty acids on a glycerol backbone inside an adipocyte. Based on these experiments, which used a similar* tracer method to the one used by Hellerstein et al. to evaluate DNL, the authors (which included Rudy Leibel, the co-discoverer of leptin) suggested that RE requires an intermediate step outside of the adipocyte in the interstitial and capillary space (figure 8 of the paper demonstrates this very well schematically).
(*) Technically, Hellerstein et al. used a heavy isotope; Leibel et al. used radioactive isotopes.
Output: Lipolysis, or “L” – Finally, in a state of fat balance, lipolysis must be equal to the sum of DNL and RE. This is true if we are talking about tiny little fat cells or giant ones. Remember, it’s the balance that matters.
I hope it’s clear from this summary that there are an infinite number of physiologic states that can satisfy the equation of fat balance: DNL + RE = L. For example, someone like me who is in fat balance (i.e., I’m neither gaining nor losing fat mass at this point) on a ketogenic diet with daily fat intake often exceeding 400 grams, has virtually zero DNL, but quite high RE, especially after meals. Consequently, I have very high L. If you took a person on a very low-fat diet (e.g., 20% fat, but 65% CHO), they would have modest DNL and low RE, but they would have low L. We would both be in fat balance, but we satisfy the equation DNL + RE = L by very different means.
OK, so let’s turn our attention to the non-equilibrium states: Net fat influx and net fat efflux.
Fat influx
In a state of net fat influx – accumulation of fat within a fat cell – the following condition must be met (on average): DNL + RE > L. (I say “on average” because, of course, a fat cell is a dynamic system with constant changes in these parameters. So, at any moment in time the balance can shift, but over a period of time the equation is correct.)
The next (overly simplistic) figure below gives you a representative state of what fat influx or ‘positive fat flux’ probably looks like. DNL is higher, but still relatively small, unless overfeeding CHO. RE is larger than it was in a balanced state, but not necessarily ‘huge.’ Most cases of net fat influx are probably governed by low L. In other words, fat accumulation is probably more governed by a failure to mobilize (breakdown TG into fatty acids for export and use) TAG than anything else.
Have you ever spoken with someone who is trying desperately to lose weight (fat) who says, “I don’t understand what’s happening…I hardly eat any fat, and yet I can’t lose a pound (of fat)!” The skinny people in the group scoff, right? Well, not so fast. It’s quite possible, if the hormones that regulate fat tissue are not working in your favor, to do such a poor job mobilizing fat from fat cells, and oxidizing that fat (see below), that you can be in fat balance, or even fat imbalance with accumulation, despite small DNL and small RE.
If you think about it, lipolysis (L), or liberating fat from a fat cell is a necessary, but not sufficient condition to actually generate the free energy inherent or stored within it. One more major step is necessary – oxidizing the fatty acid via the process of beta-oxidation. This is where one actually gets the energy (ATP) from fatty acids. The same hormones and enzymes that promote L, directly or indirectly act on other intermediaries that promote oxidation, more or less. The converse is also largely true.
Brief digression: I’m always troubled by folks who have never tried to take care of someone who is struggling to lose weight (fat), and who themselves have never been overweight, but who insist obesity is ‘simply’ an energy balance problem – people eat too many calories. When eternally lean people preach about the virtues of their ‘obvious’ solutions to obesity – just eat less and exercise more – I’m reminded of a quote (source unknown to me), “He was born on the finish line, so he thinks he won the race.” You only need to meet one woman with PCOS, or one person with hypothyroidism, or one child with Cushing’s disease to know that adiposity can – and is – largely regulated by hormones. The fact that such patients need to create a positive energy balance (i.e., eat more calories than they expend) to allow it does not seem to provide a meaningful insight into the mechanism of why.
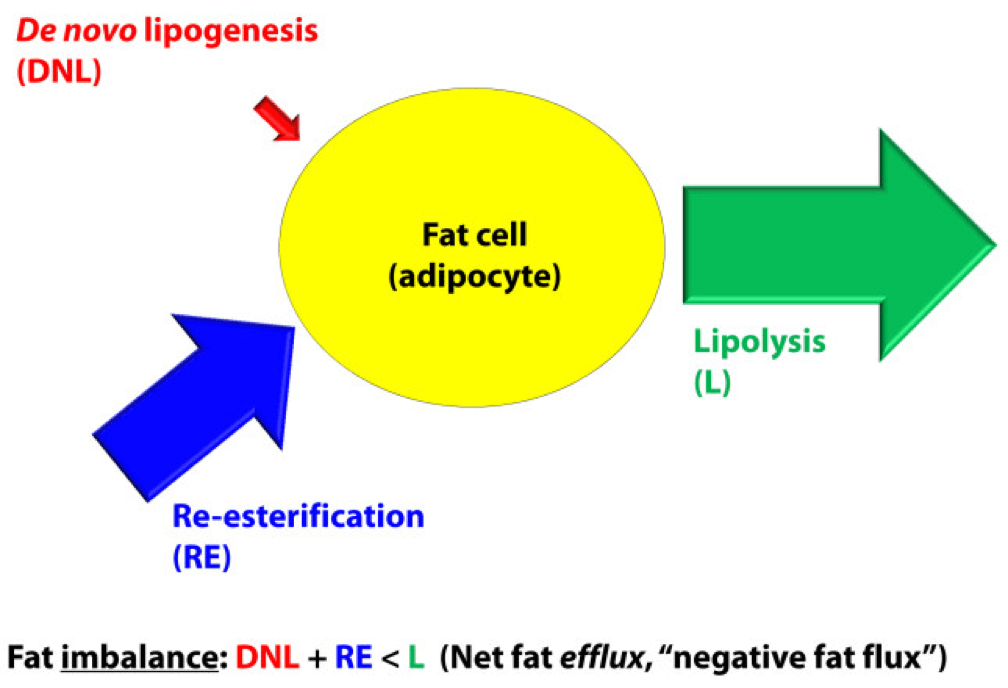
Fat efflux
In a state of net fat efflux – reduction of fat within a fat cell – the following condition must be met (on average): DNL + RE < L (same caveat as above on the idea of “on average”). Again, looking at the figure, you can see one physiologically common way this occurs, the setting of carbohydrate restriction. DNL is reduced (probably even to immeasurable levels, depending on the extent of restriction), but RE actually goes up. The net efflux, however, results from the greater increase in L.
A person in nutritional ketosis, if experiencing fat loss, probably looks like this. (Don’t worry, I have not forgot the opening questions: Does being in nutritional ketosis automatically put you in this state?). Certainly another state of net fat efflux is starvation. DNL and RE are both very small, especially DNL, and lipolysis is quite large. This is probably the most rapid state of negative fat flux a human can experience.
So what we do about it?
I do not believe there is only one state, shy of total starvation, which will assuredly put you in state of negative fat flux. Of course, starvation is not sustainable, and therefore should be taken off the table as a viable long term eating strategy.
What about profound caloric restriction? Yup, this is probably (though not necessarily) going to work, depending on how “profound” is defined. If defined as a 40% reduction of energy stable intake, it’s probably going to work. If defined as a 10% reduction, it would be difficult to know without knowing at least two other things:
- Baseline level of insulin resistance;
- RQ of pre- and post-diet.
What about dramatic alterations in macronutrient composition? This is where the discussion gets really interesting. Many people, myself included, advocate a diet that overall reduces insulin secretion. The rationale, of course, is provided by the first figure above (from the textbook) and a slew of clinical studies which I will not review here (see Gardner JAMA 2007, Ludwig JAMA 2012, and Shai NEJM 2008 to name a few).
But, the bigger question is why? Why do most (but not all, by the way) people with excess fat to spare who are on well-formulated carbohydrate-reduced diets lose fat? (Notice, I did not say weight, because the initial – and often rapid — weight loss achieved by many is actually water loss.)
Is it because of a physiologic change that leads them to reduce overall intake?
Is it because of a physiologic change that, despite the same intake in overall calories, increases their energy expenditure?
Is it some combination of these?
I wish I knew the answer, but I don’t (universally). I believe we will know the answer to this question in a few years, but until then, I’m left to offer the best my limited intuition can offer. In other words, what I suggest below is my best interpretation of the literature, my personal experience that I’ve had with hundreds of other people, and my discussions with some of the most thoughtful scientists in the world on this topic:
Thought #1: I suspect that many people who reduce simple carbohydrates and sugars end up eating fewer calories. This observation, however, may confound our understanding of why they lose weight. Do they lose weight because they eat less? Or, do they eat less because they are losing weight? I suspect the later. In this state, lipolysis — and by extension, given the hormonal milieu, oxidation — are very high, certainly relative to their previous state. By definition, L > DNL + RE, so there is ample ATP generated by oxidation of the fatty acid. If you believe (as I do*) that the liver is the master organ of appetite regulation, increases in ‘available energy’ (i.e., ATP) would naturally reduce appetite (though I don’t think we know if ATP per se is the driver of this feedback loop). But don’t confuse what’s happening. They are not giving up fat from their fat cells because they are eating less. They are eating less because they are giving up fat from their fat cells. Big difference.
(*) The especially astute reader will note that this is the first time I have made reference to this point. I have been heavily influenced recently by the work of Mark Friedman, and a discussion of this point is worth an entire post, which I promise to deliver at some point in the future. If you can’t wait, which I can understand, I highly encourage you to start scouring the literature for Mark’s work. It’s simply remarkable.
Thought #2: I also suspect that some fraction of people who follow this eating strategy lose fat without any appreciable reduction in their total caloric intake, at least initially. What?, you say, doesn’t this violate the First Law of Thermodynamics? Not at all. If L > DNL + RE, and the increase in lipolysis (i.e., fatty acid flux out of the fat cell) results in increased oxidation of fatty acids, energy expenditure (EE) would be expected to rise. A rise in EE, in the face of constant input, is a sign of fat loss. What differentiates those in this camp (I was in this camp) from those above (point #1), is unclear to me. It may have to do with concomitant exercise. I have seen unpublished data, which I can’t share, suggesting non-deliberate EE rises more in a low RQ (high fat, low carb) environment when a person is exercising significantly. I’m not stating the obvious – that the deliberate EE is higher – that is clearly true. I’m suggesting resting EE is for some reason more likely to rise in this setting. Since I’m taking the liberty of hypothesizing, I would guess this effect (if real) is a result of the body trying to keep up with a higher energy demand and making one trade-off (generating more free ATP via more lipolysis) for another (ensuring a constant supply of available energy to meet frequent demands). It is also possible that this increase in free/available energy results in an increase in deliberate EE (i.e., the person who suddenly, in the presence of a cleaned up diet feels the desire to walk up the stairs when they previously took the elevator). Finally, and perhaps most importantly, whether or not this up-regulation of energy takes place may be dependent on the other hormones in the body that also play a role in fat regulation, including cortisol, testosterone, and estrogen. They could be partly or mostly responsible for this. The literature is quite dilute with respect to this question, but in my experience (feel free to dismiss), it is not uncommon to see a reduction in cortisol and an increase in testosterone (I experienced about 50% in free and total testosterone) with a dietary shift that improves food quality. The same may be true of estrogen in women, by the way, though I have less clinical experience with estrogen.
Thought #3: As a subset to the point above (point #2), in an ‘extreme’ state of carbohydrate restriction, i.e., — nutritional ketosis — there is an energy cost of making the ketones from fatty acids. I referred to this as the “Hall Paradox” after Kevin Hall, who first alerted me to this, in this post (near the bottom of the post). What is not clear (to me, at least) is if this effect is transient and if so, how significant it is. I recall that during the first three months of my foray into nutritional ketosis, I was eating between 4,000 and 4,500 kcal/day for a 12-week period, yet my weight reduced from 176 lb (about 9.5% bf by DEXA) to 171 lb (about 7.5% bf by DEXA), which means that of the 5 pounds I lost in 12 weeks, 4 were fat tissue. Today, however, I don’t consume this much, closer to 3,800 kcal/day, and one reason may be that two years later my body is more efficient at making ketones and this so-called “metabolic advantage” is no longer present.
(I have always found the term “metabolic advantage” to be misleading, though I’m guilty of using it periodically. It’s really a metabolic disadvantage if your body requires more energy to do the same work, but nevertheless, people refer to – and argue vehemently about – this phenomenon. The question is not, does it exist? One look at individual summary data from David Ludwig’s JAMA paper on this topic makes that clear. The questions are, why does it only exist in some people, what relevance does it have to fat loss – is it cause or effect? – and, for how long does it persist?)
Thought #4: For reasons I have yet to fully understand, some people can only lose fat on a diet that restricts fat (and by extension a diet that is still high in carbohydrate, since I’m excluding starvation and profound caloric restriction from this discussion). In my experience (and Gardner’s A TO Z trial seems to validate this, at least in pre-menopausal women), about 20% of people aspiring to reduce adiposity seem to do it better in a higher RQ environment. Using the Ornish diet as the example from this paper, I suspect the reason is multifactorial. For example, the Ornish diet restricts many things, besides fat. It restricts sugar, flour, and processed carbohydrates. Much of the carbohydrate in this diet is very low in glycemic index and comes primarily from vegetables. So, I don’t really know how likely it is to lose weight on a eucaloric diet that is 60% CHO and 20% fat, if the quality of the carbohydrates is very poor (e.g., cookies, potato chips). The big confounder in these observations is that most low-fat diets, though still modestly high in RQ relative to a low-carb diet, reduce greatly the glycemic index and glycemic load, as well as the fructose.
Which brings us to the point…
Does being in nutritional ketosis ensure negative fat flux (i.e., fat loss, or L > DNL + RE)?
Being in ketosis tells us nothing about this equation! Let me repeat this: It is metaphysically impossible to infer from a measurement of B-OHB in the blood if this equation is being satisfied. It just tells us that our body is using some fraction of our dietary fat and stored fat (once it undergoes lipolysis) to make ketones, given that glucose intake is very low and protein intake is modest (net effect = minimal insulin secretion).
If you look at the figure below, you see this point (It’s simplified, obviously, and for example, does not show that fat from fat cells can be used directly by skeletal muscles). Nothing in this figure implies a reduction in the size of the cells at the bottom right of the figure. It’s quite possible, of course, since ketosis results in a large L and implies a very small DNL. But, if (small) DNL + (very large) RE is greater than (large) L, guess what? Fat flux is net positive. Fat is gained, not lost. Still in ketosis, by the way (quantified loosely by fasting levels of B-OHB greater than about 0.5 to 1 mM), but not losing fat. (I hope the first attempt at a solution in this setting is obvious by now, notwithstanding the fact that I’ve seen this situation dozens of times with more than one solution, including the ‘obvious’ one — reducing fat intake.)
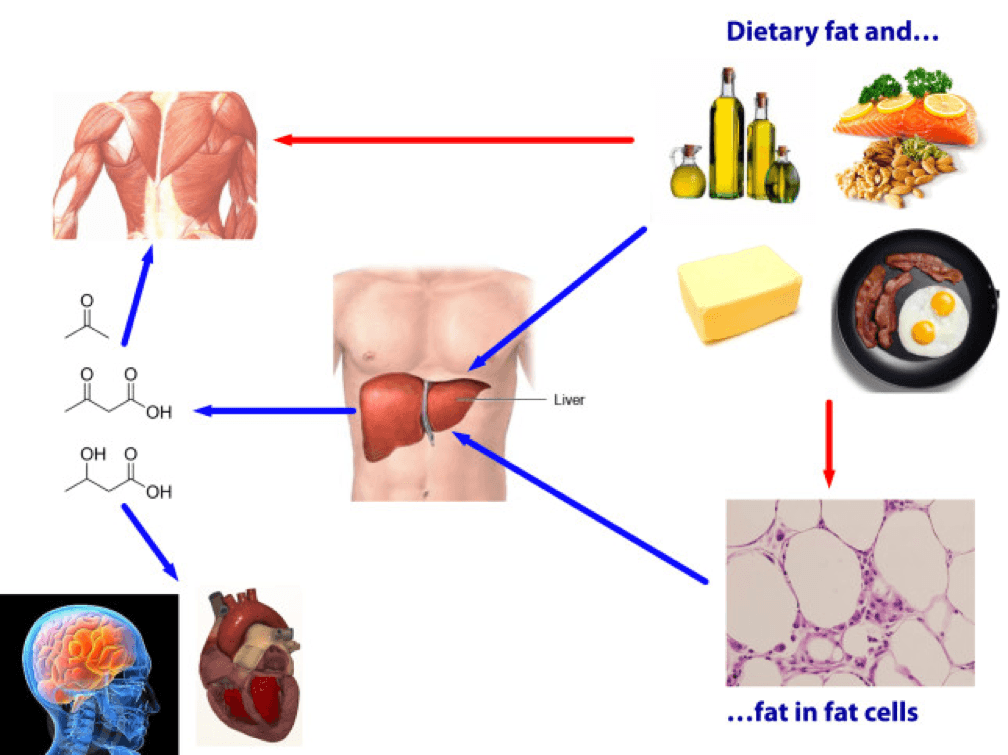
The other myth worth addressing is that the higher the level of B-OHB, the more “fat burning” that is going on. This is not necessarily true at all. As you can tell, I love equations, so consider this one:
B-OHB (measured in blood) = B-OHB produced (from dietary fat) plus B-OHB produced (from lipolysis of TAG) less B-OHB consumed by working muscles, heart, brain.
How does knowing one of these numbers (B-OHB measured in blood) give definitive answers to another (B-OHB produced from lipolysis of TAG)? It can’t. That’s the problem with multivariate algebra (and physiology).
Many people who enter nutritional ketosis do so, I worry, because they believe it “guarantees” fat loss. I hope I have convinced you that this is not true. Nutritional ketosis is one eating strategy to facilitate negative fat flux, and it works very well if done correctly. It comes with some advantages and some disadvantages, just like other eating strategies. When I get back to the series on ketosis, I will address these, but for now I felt it was very important to put things in perspective a bit. Furthermore, I am convinced that it is not the ideal eating strategy for everyone.
Delorean by Marci Maleski is licensed under CC by 2.0



Hi Peter,
1. In a low carb / ketogenic setting if one is attempting to lose weight and does not feel hungry do they need to be concerned about caloric restriction and consequent hpothyroid/down regulation of metabolism? Do they use hunger as a guide or kind of force themselves to eat enough calories?
2. Apart from reducing fat intake and addressing hormonal issues is there anything else one can try to accelerate fat oxidation? For example low intensity exercise or intermittent fasting?
3. The fat to be reduced: is it specific as SAFs or MUFAs?
Thanks
1. Hunger is probably the best guide
2. Hormones rule
3. Depends on dietary intake, but ideally, it’s the storage form of triglyceride
Do you have any recommendations for where we can read up on #3 – what you mean by storage form of TAG and how dietary intake will affect that, beyond the standard divisions of SFA, MUFA and PUFA? Or what terms to search for to find it?
Hi Peter
Please could you elaborate on why so many people have such positive results in reducing body fat while under a ketogenic diet?
Reading your previous articles they sounded like they were in favor of ketosis (atkins diet etc) yet when I read this article it seems against it?
Surely on a low carb, high fat diet once the body utilizes fats as its primary source of energy your fat stores begin to deplite? If this is not due to a ketogenic diet how else can this be achieved when under ‘normal’ diet with carbs being primary source they (glycogen)are utilized first and then once they are depleted your body uses a mixture of muscle mass and fat which is far from ideal?
Not sure why you’re interpreting this article that way. This post is simply a statement of facts. No judgement or emotion intended.
>They are not giving up fat from their fat cells because they are eating less. They are eating less because they are giving up fat from their fat cells. Big difference.
This leaves me confused as the two activities are very different. You can chose to eat less, but you cannot simply choose to give up fat from your fat cells (note: if you can, I’m impressed). The way these sentences are written imply that the only way anyone will be able to eat less is if they are already giving up fat from their fat cells. Clearly, I’m missing something.
Your understanding is likely correct over the long run. If a fat cell is “hanging on” to fat, as far as the body (esp. the liver) “knows” you are starving, and appetite goes up. One can only fight this for so long.
Hi Peter,
I really like your blog and I was hoping you could please tell me something about this ketosis mystery. My question may sound silly to you since I am not from medical background but I am really interested to know this and no one on Internet seems to have an answer for this..
My question is –
When you are in Ketosis, i understand that it is the process of your liver turning fat into ketone bodies. I fail to understand one key point is that these ketones are made up of my own body fat or the high amount of fat i am eating while on low card diet. If ketones are made up of my own body fat, why do i need to consume so high amount of fat in the first place? What happends to dietry Fat then? Is eating high amount of fat really neccesary to induce ketosis?
Made from both, but to answer your question, if you stop eating, just from your body — this is called starvation ketosis.
At some point you’ll have used so much body fat that you need to eat some fat. Having been very underweight I can assure that you can’t live on your stored body fat when you’re that low in body fat percentage despite people claiming that I’m still carrying 30,000-40,000 calories of fat.
For those interested, here is the obit for Dr. Coleman, co-discoverer of the genetic leptin role in weight:
https://www.nytimes.com/2014/04/26/us/douglas-l-coleman-82-dies-found-a-genetic-cause-of-obesity.html?ref=obituaries
Hi Peter,
1. If the brain needs 120-150 grams of carbs how can it run optimally out of ketosis at a lower number of carbs?
2. For how long generally does the insulin stay raised after eating carbs?
3. Is it better to restrict carbs to one meal or spread them over 3 meals in terms of their effect on insulin?
Thanks
1. Ketones offset this need. That’s the point.
2. Depends on the person and the carb source
3. “better?”
Hi Norm,
I am far from being a MD, but from what I have read:
1) Your brain does NOT need 120-150g of carbs. It needs a few grams but it can also use ketones.
Even if our brain couldn’t use ketones our liver can create glucose from fats: in other words you don’t need 150g of dietary carbs even if your brain where unable of using ketones.
Question 3) is insteresting. Is the peak value of glucose/insulin what causes the damage or is it the AUC the important factor and several meals can cause the same effect? Is there a glucose/insulin threshold it is better not to reach or does their effect follow a smooth curve? Intuitively spreading carbs in several meals should be better or at least equal than eating all of them in one meal: the more stable your blood glucose, the better.
Sorry Peter, I was menat to ask if restricting carbs to one meal was better than spreading them over 3 meals in terms of insulin secretion. Thanks
Hi Peter,
What do you think about ASP (Acylation Stimulating Protein) which apparently stores fat without raised insulin? Could this explain weight gain/stall on a LCHF diet for some people?
Thanks
Sorted. Gary kindly pointed me to the email exchange between Keith Frayn and Fred Hahn over ASP.
Hi! My name is Marco and i am an student 22 years of age, living in Sweden. I was wondering on your take on Bioenergetics and their take on HFLC. They smash the idea of HFLC here ->
https://www.abioenergeticview.com/2-2
Do you think it is pseudoscience when it comes to biological matter where endokrinology explains things better? I am freaked out of the debate since i wanted to pursue biochemistry for their authenticity to explaining everything on a micro scale, all the way from the cell. I thought that was were the explanations could be found for solving huge problems in the world. Do you recommend studying something else or are their take on HFLC somewhat valid?
“Eating a high-fat low-carb diet (HFLC) tends to increase levels of free fatty acids (or non-esterified fatty acids, NEFA) in the blood. While this is usually the goal of any HFLC diet, it mimics the “stress”metabolism seen in those with diabetes, obesity and old age (Frayn K, et al. 1996). However, insulin resistance isn’t necessarily induced on a hypocaloric HFLC diet because fatty acid oxidation “keeps up” with fatty acid mobilization.Free fatty acids suppress mitochondrial respiration (Kamikawa and Yamakazi, 1981), leading to increased glycolysis (and the production of lactic acid) to maintain cellular energy.
Lactic acid along with the longer chain fatty acids inhibit the regulatory enzyme pyruvate dehydrogenase (Bradley N, et al. 2008), which is activated by insulin and links the metabolism of glucose via glycolysis with the Krebs cycle in the mitochondria. The inactivation of pyruvate dehydrogenase (PDH) reduces the production of carbon dioxide (increasing lactic acid through lactate dehydrogenase) whereas carbohydrates have the opposite effect.
Marco, are you sure that the words in the link you posted are not placed at random?
Nevertheles, let’s assume they are not and let’s talk about science:
1) Make a guess: “HFLC diets support mitochondrial dysfunction”
2) Make a prediction (?)
3) Design an experiment to test your prediction (?)
4) Analyze the results (?)
Or it seems endokrinology as of Ray Peat among others also support glukose as healthy and the PUFA’s more dangerous in the equation.
“If Dr. Budd’s thinking (and results) had been more widely accepted when his publications appeared, thinking about “diabetes” might have led to earlier investigation of the syndromes of stress and tissue wasting, with insulin being identified as just one of many regulatory substances, and a large amount of useless and harmful activity treating hyperglycemia as the enemy, rather than part of an adaptive reaction, might have been avoided.”
https://raypeat.com/articles/articles/glucose-sucrose-diabetes.shtml
Here comes the last quote – sorry spamming Peter but i am intrigued to see if you done simplicity to early and reductionism to a hard complex biological matter question.
Carbs are needed to activate inactived T4 thyroid gland and much much more. Excessive Triglycerides from eating bad fats that have been oxidised which many overdo on HFLC or to much SFA and MCT contribute to over inflammation in the body. When you then enter ketosis, you enter a slower metabolism as fat burns slower than glycogen. Say goodbye to performing HIIT, Crossfit, Soccer and many more activities that actually build type IIb fiber muscles that keep you from storing fat, while builds muscle when you have glycogen levels intact and fueled.
I love intermittent fasting with fats such as coconut and macademia nuts but to much fat in the body can mimic the exposure under training with triglycerides, the same oxidative way like A.G:E products?
If you do intermittent fasting with cooked greens in blender with fats and berries (they say not to mix fats with sugar but you can ferment the berries with salt and lemon to reduce sugars) is prone to be beneficial to balance thyroids and limit methionine and triglycerides oxidative processes i think, cause you only refeed fats on fasting days. Carbs and protein on training days, fats on IF days. What do you think about that?
Low carb / ketogenic eaters have a normal blood sugar, so your hypothesis needs to explain why things like thyroid function depend on ingested carbohydrate as opposed to the endogenous glucose available in nutritional ketosis.
Vincente, i am not qualified to engage in a personal experiment cause i can’t on a cellular level, test for true causality. These experts which studies i linked at no random place, was given reference to my concern with LCHF, as i adressed to peter and everybody on the blog. My take is ” moderate carbs, moderate fats on fasting days with 2 meals to not overeat and indulge in stimulating the sensitive actions of insulin” and everyother training day to have “moderate protein, moderate carbs with very little fat”. A refeed strategy for energy needs but overall health by balance conflicting theories of food intake. Moderation as Peter, Ray Peat or any other expert out there, can make statements now that will be devestating when new science paradigm enters later on. I think the paradigm will come when one is an expert of general science in every field – if one has the abillity to study biochemistry, endokrinology, neuro etc. etc. etc. Every studyfield shows different results by huge standards.
The authors of the article are “experts”? I don’t believe that. Their reasoning is so poor…
Just a couple of examples: they talk about miochondrial energy efficiency and you can’t find the word “ketone” in their speculations.
Another one, they say that “consuming a HFLC diet mimics a diabetic’s metabolism” so as long as “an increase in lactic acid is a common feature of diabetes”, HFLC diets are energetically inefficient and “when energy production becomes “inefficient” reactive oxygen species (ROS) are produced in large amounts”. Even if that speculation were true, the reasoning seems BS to me.
IMHO, a piece of garbage.
Thanks for your comment Vincent. You seem to reason well and question things well. They are acclaimed experts, at least in the public eye but i may need more scientific approach to myself question these claims. As of now, i don’t so thanks for contributing Vincente! 🙂
As a thank you gift, i would like to share this medicinal context for how to lower risks with LCHF with spices for (in case Attia and researchers in the same field have gotten LCHF wrong). This study is done on overweight but otherwise “healthy” people so may not be the best chosen group as they are likely to already have high drawing abbillities for triglycerides i suppose. But anyhow:
“According to Penn State researchers, eating a diet rich in spices, like turmeric and cinnamon, reduces the body’s negative responses to eating high-fat meals. Sheila West, associate professor of biobehavioral health, Penn State, who led the study said that people eating a high-fat meal end up with high levels of triglycerides (a type of fat) in their blood.We found that adding spices to a high-fat meal reduced triglyceride response by about 30 percent, compared to a similar meal with no spices added.”
I should add that bioperine from black pepper, oils and cooking activates a far more bioavailable tumeric.
Cinnamon i prefer to add to raw hot chocolate with goat milk (just slow-boil the milk seperate). Have a great day!
“Sheila West, associate professor of biobehavioral health, Penn State, who led the study said that people eating a high-fat meal end up with high levels of triglycerides (a type of fat) in their blood”.
Low-carb high-fat diets do NOT raise triglycerides. Read “Systematic review and meta-analysis of clinical trials of the effects of low carbohydrate diets on cardiovascular risk factors”. Obes Rev. 2012 Nov;13(11):1048-66. Epub 2012 Aug 21″.
She is probably not wired to think scientifically.
Wow, thanks for the reference. I guess we really need Peter and you Vincente and that mindset to set an end to flaw-based statements. You all really motivates me to study really hard now and ask the right kind of questions in research later in life, by involing more data before statements are made in my limited field of research…
Have a great day!
Hi Marco,
Peter is a MD and a nutrition expert. I am neither of the two.
You don’t need me, specially since my personal experience (LCHF diet has made wonders in my quality of life) makes me a clearly biased source. Read the scientific articles that report clinical trials and intervention studies in humans (I would ignore other kinds of scientific articles) and choose what you think is best for you.
Have a nice day
https://www.abioenergeticview.com/2-3 I debunk this if nutritonal ketoss is done right but i do have a question about the gains of nutritonal ketosis in testo production…
I get this in my mailbox (a series of bioenergetic views on LCHF). Peter take the BIG picture in the whole of his nutritonal ketosis which i don’t see them doing above. Peters diet is healthy as it includes low methionine content indirectly by a low protein diet (excessive methionine increase R.O.S and the “fight or fly” exhaustive stress gene. But from a nutritional standpoint in regards to testo production, the IGF-1 are clearly raised by fatty acid consumption as free testosteron is excreded right?
That is good for balancing Intermittent fasting that induces IGF-1 and autoghapy which then rejuvenates old damaged neuron cells, and cells in the mitocondria. But does eating much fat – Nutritonal ketogenic fasting – increase autoghapy for the worse and inhibit the induced IGF-1 by increasing free testo?
I ask cause i want to limit testo as much as possible and gain complex of female attributes regarding muscle. They have thinner, longer and a more elegant storage of the muscles – they dont gain as much volume. I have trained for fast muscle fibers and heavy weight training with 55kilo weighted Dips, 150 kilo squat before i accumulated damage and injured my leg from bad standpoint both execution wize and inflammatory rich diet. I now have lost that huge volume of my body but my arms are still big enough compared to girls. I weight like 68 kilo and am 1.80 cm. I think i have the wrong muscle fibers that do benefit skeletal but not my identity esthetically. For my goal of being more thin in muscle volume with a balance of 1 day HIIT, 1 day really high reps strenght training with low weights, and one day to to calisthetics for 20 minutes, and approach lower testo by:
Lose IGF-1 one day with fasting and lower fats and benefit autoghapy for HEALTH and Longevity
Increase IGF-1 the other day by GOAT colostrum powder?
https://www.scientificphysicaltherapy.com/SPT%2019_3_2010.pdf
The article discusses IGF-1 as a marker increased only by short high intensity training. And i know Peter performs at this levels sometimes besides longer marathon training. But isn’t a shorter lifespan with much higher life quality prefered over induced IGF-1 longevity by diet, excercise and from the AGE DHEA & TESTO DECLINED after 25 with skeletal loss after age 30? And if the answer is yes for you, then is LCHF really the ideal for HIIT and heavy weight training? I know that if you wait for insulin responses with the postworkout meal, excluding carbs, the skeletal and hypertrofy can benefit even more according to some research, including Mercolas. But for the thyroid gland and recovery overall, is not overall some carbs from blueberries, sea buckthorn berries with omega 7 and lastly, goat milk or sheep milk with folate and b12 and taurine rich, prefered benefits to neurological health and thyroid health, over LCHF all the time? Can’t one switch to heavy training with Soccer HIITS and carb rebounds 80g 3hours after, and 40g at night to stabilise blood sugar, and the next day, get into intermittent fasting with coconut milk and organic cooked + juiced greens along that?
Can’t one alternate or does this induce getting into NK several days?
“Summary:
The growth factor IGF-1 and the anabolic hormones growth hormone and testosterone stimulates
protein synthesis and tissue regeneration. IGF-1 can be decreased by mild aerobic exercise at the la
ctate
threshold which is approximately 50% VO2 max. This would be beneficial in cases where the patient has
increased risk of cancer. Exercising at levels higher than 60% VO 2 max will increase IGF-1 and high resistance exercise will also increase IGF-1. Growth hormones are stimulated by high intensity exercise both short sprint exercise and resistance exercise if it provides high tissue stress.
Resistance exercises with high workloads are the most effective way to stimulate the release of test
osterone. Clinically it may look like IGF-1, growth hormones and testosterone can only be stimulated later in
the treatment program when the patient has progressed into higher resistance exercise protocols”.
You know what – let me simplify: I wanted to raise IGF-1 without testosterone in order to gain a more female skinny complex. This cannot apparently be done but i can offset chronically high testo production and balance my lifestyle so i have a higher chance. I think IGF-1 is needed to be raised in order to achieve that fat loss, but reduced type II fast and heavy weight training, is i think, required for not bulking up volume when having raised IGF-1. IGF-1 is still highly benefical for my goal as it helps collagen, firmness, greater fat loss, etc. etc.
I wanted to eat carbs 3hours after training and late at night – this spikes the insuline somewhat and apparently, the IGF-1 is at the highest the first 1-2 hours during sleep. So i thought i could stabilise my blood sugar by eating carbs late at night and yes, this does that but also induces IGF-1. I thought the body handled fat very poorly at night but now i see in context of IGF-1 and satiety for better sleep, why Peter and many is recommending fat at night (disclaimer for peter: i don’t know for sure his recommendations but i THINK it was similar to what i wrote above).
So a traing with 50% vo2 max which induces IGF-1, and decreased dietary fat & protein which induce IGF-1 and testo, with some days of explosive soccer HIIT to increase IGF-1, with dietary Goat colostrum powder, more protein and dietary fats to increase IGF-1. I think it is the best way to get lean in a “skinnier” way as a guy. A chronically high testo and muscle volume is what i am seeking to exclude. I wrote this in case anyone can benefit from my reasoning. Sorry if being somewhat Off Topic!
Another post but i am so motivated so hope this come across. Peter Attia, have you considered the negative side effects of overconsuming SCA and MCT fats in your NK diet because they lead to 30% higher needs of Choline, than a dietary moderate PUFA-diet? There are over 69 found Phosphoslipids in Krill Oil but still you supplement Carlsons liquid fish oil as you stated in an interview, overloading triglyceride converting-work in the liver?
I recommend that you investigate in Choline further for pregnancies if you can because your credentials and position at the moment in the public eye, can really draw much needed attention to global problems of ADD, ADHD, poor study results, lower focus abbillities, depression, suicides etc. – all derived from Choline deficiency and probably overeating MCT’s by thus increasing the needs of Choline. Apparently, the need of 550mg daily as a male, is just a guess and hypothesised of being to small so this is already an issue with vegans and non-egg or liver eaters.
Choline is what really keep cortisol markers away well as depression and anxiety. It also regulates the metabolism and hinders acumulating a fatty-liver profoundly according to many resource studies found by googling. The daily Lipid Blog is one of them. Mercola states that “Prior research has concluded that choline intake during pregnancy “super-charged” the brain activity of animals in utero, indicating that it may boost cognitive function, improve learning and memory, and even diminish age-related memory decline and the brain’s vulnerability to toxins during childhood, as well as conferring protection later in life1.
Interestingly, the higher choline intake led to changes in epigenetic markers in the fetus. Specifically, it affected markers that regulate the hypothalamic-pituitary-adrenal (HPA) axis, which controls hormone production and activity. The higher intake of choline contributed to a more stable HPA axis, which in turn meant lower cortisol levels in the fetus. The changes in fetal genetic expression will likely continue into adulthood, where they play a role in disease prevention”.
Epigenetic Changes May Last for Generations / https://articles.mercola.com/sites/articles/archive/2012/10/08/choline-consumption-during-pregnancy.aspx