Want to catch up with other articles from this series?
- The straight dope on cholesterol – Part I
- The straight dope on cholesterol – Part II
- The straight dope on cholesterol – Part III
- The straight dope on cholesterol – Part IV
- The straight dope on cholesterol – Part V
- The straight dope on cholesterol – Part VI
- The straight dope on cholesterol – Part VII
- The straight dope on cholesterol – Part VIII
- The straight dope on cholesterol – Part IX
Previously, in Part I, Part II and Part III of this series, we addressed these 5 concepts:
#1 — What is cholesterol?
#2 — What is the relationship between the cholesterol we eat and the cholesterol in our body?
#3 — Is cholesterol bad?
#4 — How does cholesterol move around our body?
#5 –How do we measure cholesterol?
Quick refresher on take-away points from previous posts, should you need it:
- Cholesterol is “just” another fancy organic molecule in our body but with an interesting distinction: we eat it, we make it, we store it, and we excrete it – all in different amounts.
- The pool of cholesterol in our body is essential for life. No cholesterol = no life.
- Cholesterol exists in 2 forms – unesterified or “free” (UC) and esterified (CE) – and the form determines if we can absorb it or not, or store it or not (among other things).
- Much of the cholesterol we eat is in the form of CE. It is not absorbed and is excreted by our gut (i.e., leaves our body in stool). The reason this occurs is that CE not only has to be de-esterified, but it competes for absorption with the vastly larger amounts of UC supplied by the biliary route.
- Re-absorption of the cholesterol we synthesize in our body (i.e., endogenous produced cholesterol) is the dominant source of the cholesterol in our body. That is, most of the cholesterol in our body was made by our body.
- The process of regulating cholesterol is very complex and multifaceted with multiple layers of control. I’ve only touched on the absorption side, but the synthesis side is also complex and highly regulated. You will discover that synthesis and absorption are very interrelated.
- Eating cholesterol has very little impact on the cholesterol levels in your body. This is a fact, not my opinion. Anyone who tells you different is, at best, ignorant of this topic. At worst, they are a deliberate charlatan. Years ago the Canadian Guidelines removed the limitation of dietary cholesterol. The rest of the world, especially the United States, needs to catch up. To see an important reference on this topic, please look here.
- Cholesterol and triglycerides are not soluble in plasma (i.e., they can’t dissolve in water) and are therefore said to be hydrophobic.
- To be carried anywhere in our body, say from your liver to your coronary artery, they need to be carried by a special protein-wrapped transport vessel called a lipoprotein.
- As these “ships” called lipoproteins leave the liver they undergo a process of maturation where they shed much of their triglyceride “cargo” in the form of free fatty acid, and doing so makes them smaller and richer in cholesterol.
- Special proteins, apoproteins, play an important role in moving lipoproteins around the body and facilitating their interactions with other cells. The most important of these are the apoB class, residing on VLDL, IDL, and LDL particles, and the apoA-I class, residing for the most part on the HDL particles.
- Cholesterol transport in plasma occurs in both directions, from the liver and small intestine towards the periphery and back to the liver and small intestine (the “gut”).
- The major function of the apoB-containing particles is to traffic energy (triglycerides) to muscles and phospholipids to all cells. Their cholesterol is trafficked back to the liver. The apoA-I containing particles traffic cholesterol to steroidogenic tissues, adipocytes (a storage organ for cholesterol ester) and ultimately back to the liver, gut, or steroidogenic tissue.
- All lipoproteins are part of the human lipid transportation system and work harmoniously together to efficiently traffic lipids. As you are probably starting to appreciate, the trafficking pattern is highly complex and the lipoproteins constantly exchange their core and surface lipids.
- The measurement of cholesterol has undergone a dramatic evolution over the past 70 years with technology at the heart of the advance.
- Currently, most people in the United States (and the world for that matter) undergo a “standard” lipid panel which only directly measures TC, TG, and HDL-C. LDL-C is measured or most often estimated.
- More advanced cholesterol measuring tests do exist to directly measure LDL-C (though none are standardized), along with the cholesterol content of other lipoproteins (e.g., VLDL, IDL) or lipoprotein subparticles.
- The most frequently used and guideline-recommended test that can count the number of LDL particles is either apolipoprotein B or LDL-P NMR which is part of the NMR LipoProfile. NMR can also measure the size of LDL and other lipoprotein particles, which is valuable for predicting insulin resistance in drug naïve patients (i.e., those patients not on cholesterol-lowering drugs), before changes are noted in glucose or insulin levels.
Concept #6 – How does cholesterol actually cause problems?
If you remember only one factoid from the previous three posts on this topic, remember this: If you were only “allowed” to know one metric to understand your risk of heart disease it would be the number of apoB particles (90-95% of which are LDLs) in your plasma. In practicality, there are two ways to do this:
- Directly measure (i.e., not estimate) the concentration of apoB in your plasma (several technologies and companies offer such an assay). Recall, there is one apoB molecule per particle;
- Directly measure the number of LDL particles in your plasma using NMR technology.
If this number is high, you are at risk of atherosclerosis. Everything else is secondary.
Does having lots of HDL particles help? Probably, especially if they are “functional” at carrying out reverse cholesterol transport, but it’s not clear if it matters when LDL particle count is low. In fact, while many drugs are known to increase the cholesterol content of HDL particles (i.e., HDL-C), not one to date has ever shown a benefit from doing so. Does having normal serum triglyceride levels matter? Probably, with “normal” being defined as < 70-100 mg/dL, though it’s not entirely clear this is an independent predictor of low risk. Does having a low level of LDL-C matter? Not if LDL-P (or apoB) are high, or better said, not when the two markers are discordant.
But why?
As with the previous topics in this series, this question is sufficiently complex to justify several textbooks – and it’s still not completely understood. My challenge, of course, is to convey the most important points in a fraction of that space and complexity.
Let’s focus, specifically, on the unfortunately-ubiquitous clinical condition of atherosclerosis – the accumulation of sterols and inflammatory cells within an artery wall which may (or may not) narrow the lumen of the artery.
Bonus concept: It’s important to keep in mind that this disease process is actually within the artery wall and it may or may not affect the arterial lumen, which is why angiograms can be normal in persons with advanced atherosclerosis. As plaque progresses it can encroach into the lumen leading to ischemia (i.e., lack of oxygen delivery to tissues) as the narrowing approaches 70-75%, or the plaque can rupture leading to a thrombosis: partial leading to ischemia or total leading to infarction (i.e., tissue death).
To be clear, statistically speaking, this condition (atherosclerotic induced ischemia or infarction) is the most common one that will result in the loss of your life. For most of us, atherosclerosis (most commonly within coronary arteries, but also carotid or cerebral arteries) is the leading cause of death, even ahead of all forms of cancer combined. Hence, it’s worth really understanding this problem.
In this week’s post I am going to focus exclusively on what I like to call the “jugular issue” – that is, I’m going to discuss the actual mechanism of atherosclerosis. I’m not going to discuss treatment (yet). We can’t get into treatment until we really understand the cause.
“It is in vain to speak of cures, or think of remedies, until such time as we have considered of the causes . . . cures must be imperfect, lame, and to no purpose, wherein the causes have not first been searched.”
—Robert Burton, The Anatomy of Melancholy, 1621
The sine qua non of atherosclerosis is the presence of sterols in arterial wall macrophages. Sterols are delivered to the arterial wall by the penetration of the endothelium by an apoB-containing lipoprotein, which transport the sterols. In other words, unless an apoB-containing lipoprotein particle violates the border created by an endothelium cell and the layer it protects, the media layer, there is no way atherogenesis occurs.
For now, let’s focus only on the most ubiquitous apoB-containing lipoprotein, the LDL particle. Yes, other lipoproteins also contain apoB (e.g., chylomicrons, remnant lipoproteins such as VLDL remnants, IDL and Lp(a)), but they are few in number relative to LDL particles. I will address them later.
The endothelium is the one-cell-thick-layer which lines the lumen (i.e., the “tube”) of a vessel, in this case, the artery. Since blood is in direct contact with this cell all the time, all lipoproteins – including LDL particles – come in constant contact with such cells.
So what drives an LDL particle to do something as sinister as to leave the waterway (i.e., the bloodstream) and “illegally” try to park at a dock (i.e., behind an endothelial cell)? Well, it is a gradient driven process which is why particle number is the key driving parameter.
As it turns out, this is probably a slightly less important question than the next one: what causes the LDL particle to stay there? In the parlance of our metaphor, not only do we want to know why the ship leaves the waterway to illegally park in the dock, but why does it stay parked there? This phenomenon is called “retention.”
Finally, if there was some way an LDL particle could violate the endothelium, AND be retained in the space behind the cell (away from the lumen on the side aptly called the sub-endothelial side) BUT not elicit an inflammatory (i.e., immune) response, would it matter?
I don’t know. But it seems that not long after an LDL particle gets into the sub-endothelial space and takes up “illegal” residence (i.e., binds to arterial wall proteoglycans), it is subject to oxidative forces and as one would expect an inflammatory response is initiated. The result is full blown mayhem. Immunologic gang warfare breaks out and cells called monocytes and macrophages and mast cells show up to investigate. When they arrive, and find the LDL particle, they do all they can to remove it. In some cases, when there are few LDL particles, the normal immune response is successful. But, it’s a numbers game. When LDL particle invasion becomes incessant, even if the immune cells can remove some of them, it becomes a losing proposition and the actual immune response to the initial problem becomes chronic and maladaptive and expands into the space between the endothelium and the media.
The multiple-sterol-laden macrophages or foam cells coalesce, recruit smooth muscle cells, induce microvascularization, and before you know it complex, inflamed plaque occurs. Microhemorrhages and microthrombus formations occur within the plaque. Ultimately the growing plaque invades the arterial lumen or ruptures into the lumen inducing luminal thrombosis. Direct luminal encroachment by plaque expansion or thrombus formation causes the lumen of the artery to narrow, which may or may not cause ischemia.
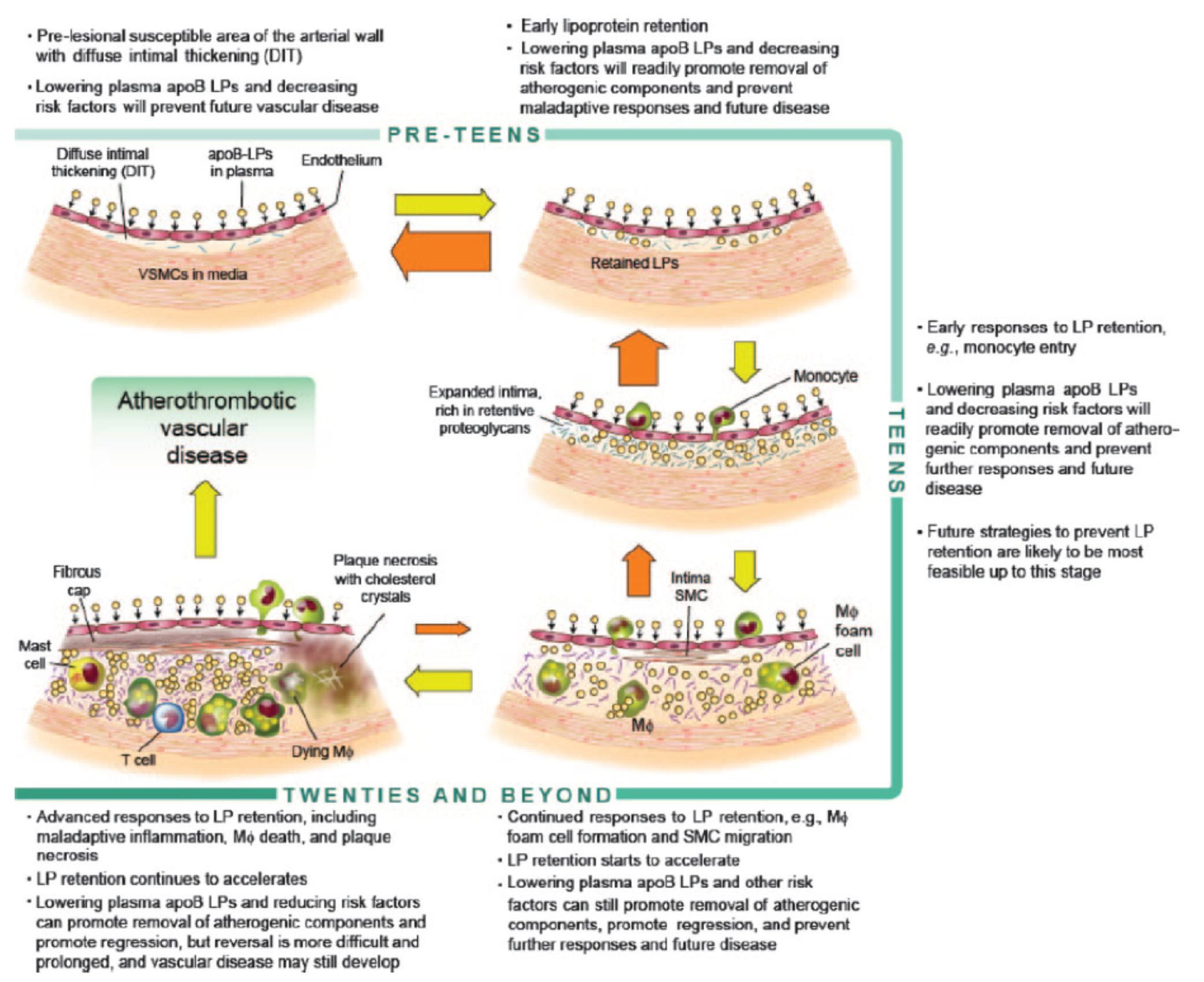
Before we go any further, take a look at the figure below from an excellent review article on this topic from the journal Circulation – Subendothelial Lipoprotein Retention as the Initiative Process in Atherosclerosis. This figure also discuss treatment strategies, but for now just focus on the pathogenesis (i.e., the cause of the problem).
In this figure you can see the progression:
- LDL particles (and a few other particles containing apoB) enter the sub-endothelium
- Some of these particles are retained, especially in areas where there is already a bit of extra space for them (called pre-lesion susceptible areas)
- “Early” immune cells initiate an inflammatory response which includes the direct interaction between the LDL particle and proteins called proteoglycans.
- The proteoglycans further shift the balance of LDL particle movement towards retention. Think of them as “cement” keeping the LDL particles and their cholesterol content in the sub-endothelial space.
- More and more apoB containing particles (i.e., LDL particles and the few other particles containing apoB) enter the sub-endothelial space and continue to be retained, due to the existing “room” being created by the immune response.
- As this process continues, an even more advanced form of immune response – mediated by cells called T-cells – leads to further retention and destruction of the artery wall.
- Eventually, not only does the lumen of the artery narrow, but a fibrous cap develops and plaques take form.
- It is most often these plaques that lead to myocardial infarction, or heart attacks, as they eventually dislodge and acutely obstruct blood flow to the portion of muscle supplied by the artery.
Another way to see this progression is shown in the figure below, which shows the histologic progression of atherosclerosis in the right coronary artery from human autopsy specimens. This figure is actually a bit confusing until you understand what you’re looking at. Each set of 3 pictures shows the same sample, but with a different kind of histological stain. Each row represents a different specimen.
- The column on the left uses a stain to highlight the distinction between the intimal and media layer of the artery call. The intima, recall, is the layer just below the endothelium and should not be as thick as shown here.
- The middle column uses a special stain to highlight the presence of lipids within the intimal layer.
- The right column uses yet a different stain to highlight the presence of macrophages in the intima and the media. Recall, macrophages are immune cells that play an important role of the inflammatory cascade leading to atherosclerosis.
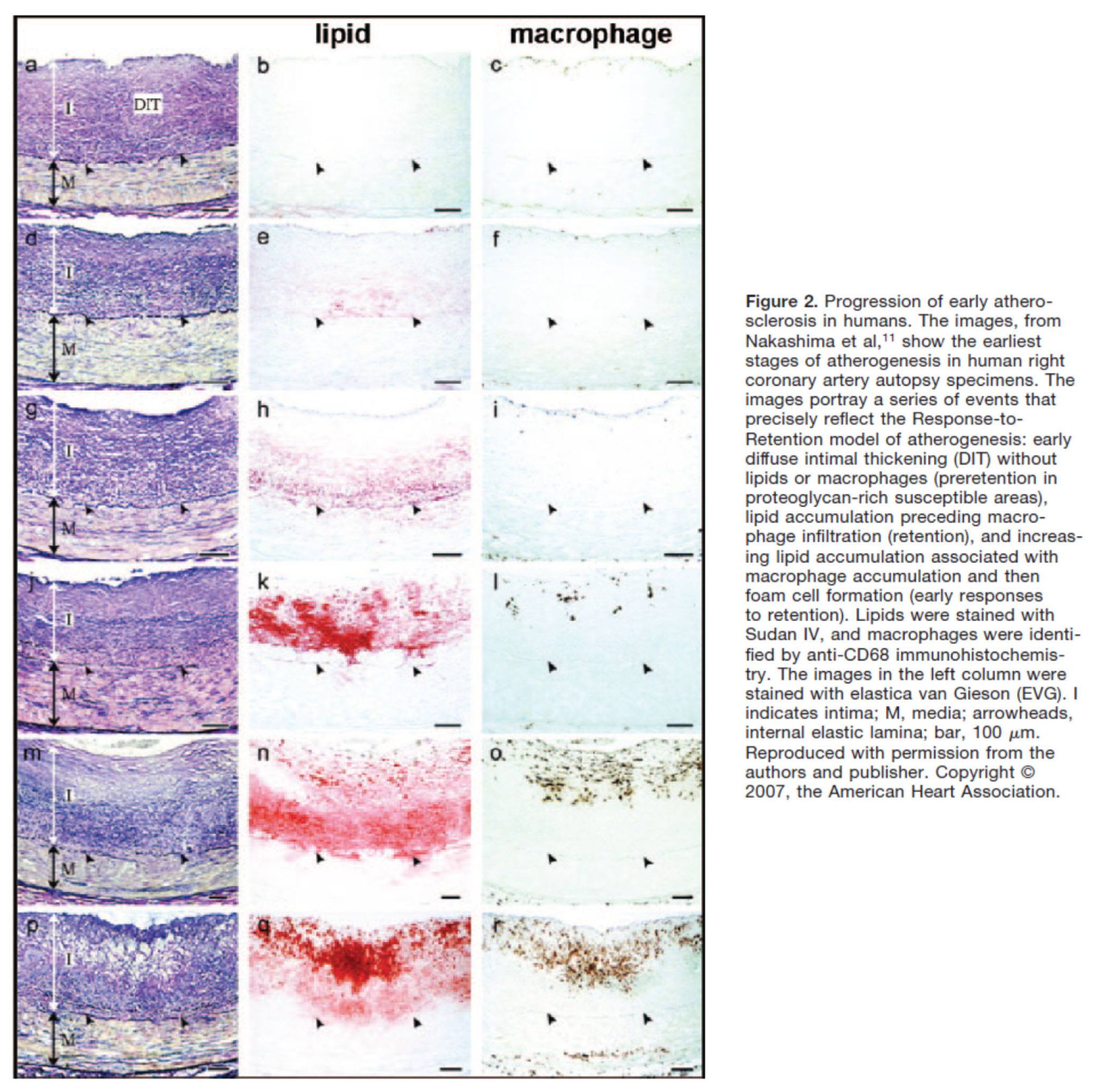
What is particularly compelling about this sequence of figures is that you can see the trigger of events from what is called diffuse intimal thickening (“DIT”), which exacerbates the retention of lipoproteins and their lipid cargo, only then to be followed by the arrival of immune cells, which ultimately lead the inflammatory changes responsible for atherosclerosis.
Why LDL-P matters most
You may be asking the chicken and egg question:
Which is the cause – the apoB containing LDL particle OR the immune cells that “overreact” to them and their lipid cargo?
You certainly wouldn’t be alone in asking this question, as many folks have debated this exact question for years. The reason, of course, it is so important to ask this question is captured by the Robert Burton quote, above. If you don’t know the cause, how can you treat the disease?
Empirically, we know that the most successful pharmacologic interventions demonstrated to reduce coronary artery disease are those that reduce LDL-P and thus delivery of sterols to the artery. (Of course, they do other things also, like lower LDL-C, and maybe even reduce inflammation.)
Perhaps more compelling is the “natural experiment” of people with genetic alterations leading to elevated or reduced LDL-P. Let’s consider an example of each:
- Cohen, et al. reported in the New England Journal of Medicine in 2006 on the cases of patients with mutations in an enzyme called proprotein convertase subtilisin type 9 or PCSK9 (try saying that 10 times fast). Normally, this proteolytic enzyme degrades LDL receptors on the liver. Patients with mutations (“nonsense mutations” to be technically correct, meaning the enzyme is somewhat less active) have less destruction of hepatic LDL receptors. Hence, they have more sustained expression of hepatic LDL receptors, improved LDL clearance from plasma and therefore fewer LDL particles. These patients have very low LDL-P and LDL-C concentrations (5-40 mg/dL) and very low incidence of heart disease. Note that a reduction in PCSK9 activity plays no role in reducing inflammation.
- Conversely, patients with familial hypercholesterolemia (known as FH) have the opposite problem. While there are several variants and causes of this disease, the common theme is having decreased clearance of apoB-containing particles, often but not always due to defective or absent LDL receptors, leading to the opposite problem from above. Namely, these patients have a higher number of circulating LDL particles, and as a result a much higher incidence of atherosclerosis.
So why does having an LDL-P of 2,000 nmol/L (95th percentile) increase the risk of atherosclerosis relative to, say, 1,000 nmol/L (20th percentile)? In the end, it’s a probabilistic game. The more particles – NOT cholesterol molecules within the particles and not the size of the LDL particles – you have, the more likely the chance a LDL-P is going to ding an endothelial cell, squeeze into the sub-endothelial space and begin the process of atherosclerosis.
What about the other apoB containing lipoproteins?
Beyond the LDL particle, other apoB-containing lipoproteins also play a role in the development of atherosclerosis, especially in an increasingly insulin resistant population like ours. In fact, there is some evidence that particle-for-particle Lp(a) is actually even more atherogenic than LDL (though we have far fewer of them). You’ll recall that Lp(a) is simply an LDL particle to which another protein called apoprotein(a) is attached, which is both a prothrombotic protein as well as a carrier of oxidized lipids – neither of which you want in a plaque. The apo(a) also retards clearance of Lp(a) thus enhancing LDL-P levels. It may foster greater penetration of the endothelium and/or greater retention within the sub-endothelial space and/or elicit an even greater immune response.
In summary
- The progression from a completely normal artery to an atherosclerotic one which may or may not be “clogged” follows a very clear path: an apoB containing particle gets past the endothelial layer into the sub-endothelial space, the particle and its cholesterol content is retained and oxidized, immune cells arrive, an initially-beneficial inflammatory response occurs that ultimately becomes maladaptive leading to complex plaque.
- While inflammation plays a key role in this process, it’s the penetration of the apoB particle, with its sterol passengers, of the endothelium and retention within the sub-endothelial space that drive the process.
- The most numerous apoB containing lipoprotein in this process is certainly the LDL particle, however Lp(a) (if present) and other apoB containing lipoproteins may play a role.
- If you want to stop atherosclerosis, you must lower the LDL particle number.
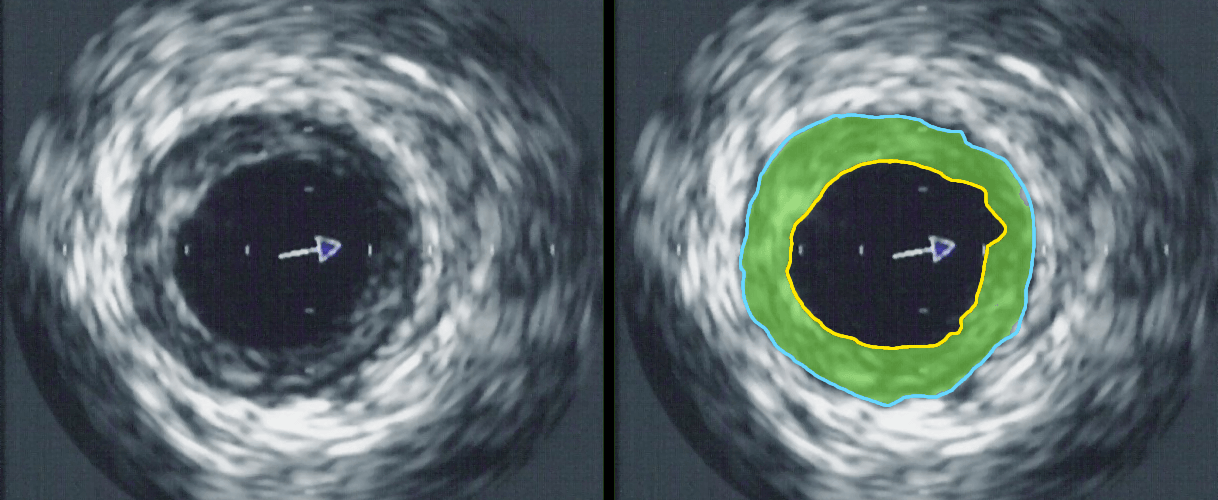
Intravascular ultrasound image of a coronary artery (left), with color coding on the right, delineating the lumen (yellow), external elastic membrane (blue) and the atherosclerotic plaque burden (green). The original uploader was Ksheka at English Wikipedia [CC BY-SA 2.5], via Wikimedia Commons

{kind=link}
First off many thanks for this excellent. detailed and extensive series.
You say “If you were only “allowed” to know one metric to understand your risk of heart disease it would be the number of apoB particles…” Can you please say what you consider to be a normal vs. high number of apoB particles?
If I may offer, it seems that there is still confusion (in some comments) as to the difference between the various LDL particle counts and what most of us see reported by the standard test which is (as I understand it) a VOLUME. So if I correctly follow your reasoning: “1,000 large LDL particles are no better or worse than 1,000 small LDL particles” BUT with the standard test 1,000 large particles would result in an higher LDL-C “number” than 1,000 small particles.
1,000 nmol/L is 20th percentie. 1,200 is about 50th, and 2,000 is about 95th percentile.
Is there a correlation between high LP(a) and high LDL-P?
Can you have very low VLDL-P and high LDL-P?
Thanks again Peter!
and also… are high LP(a) and LDL-P determined largely by genes? How far does nutrition go in lowering them?
(ignore question if this will be covered in next part) : )
Genes play a huge role, and I am not aware of the correlation between lp(a) and LDL-P. It may exist, but I’m not familiar with these data.
1. High fat diets DO raise LDL-C — let me repeat LDL-choleserol — in about a third of people.
2. This is NOT the same as raising LDL-P, and in fact many times LDL-C can go up, while LDL-P goes down for reasons I will explain in a future post.
Be interested in your further exploration of this. In my case high fat diet does raise LDL-C and LDL-P with LDL-P going through the roof! Am guessing that multiple factors at play: high absorption,genetic predisposition to lots of particles and a CETP problem TRG rich cholesterol poor LDL particles. Only thing that changes on high fat is not particle count, but mix of large and small which is irrelevant.
I believe that gradient process clearly is the issue as is the architecture of the coronary arteries. If you have lots of particles moving through areas where arteries are more narrow, ie LAD
you are likely to see more damage. Also, the health of the artery itself,particularly as one ages may be less robust( maybe emits less nitric oxide)thereby allowing particles to penetrate the wall. Infection clearly plays a part,but this appears to be after the artery wall has been penetrated with particles that have been glycated more likely to stay and turn in to a plaque instead of being carried out and returned to the liver.
CRP is not so good a marker; plenty have plaques and coronary events with low CRP.
Question: Would an atopic allergic person be more prone to CAD? Immune system already overreacts and maybe it does so when particles penetrate the artery wall.
Great series. Thanks for being so generous with your time on a complex subject
Is it really that clear that it is only LDL particle number, and not size, that matters? Intuitively, it would seem logical that it would be easier for small, than large, particles to squeeze into the sub-endothelium. Could both size and number matter? Also, I’m curious what it is about ApoB etc that apparently makes LDL particles so much more likely than HDL particles to enter the sub-endothelium.
Thanks!
We’ll cover this in great detail in one of the upcoming posts. If you want a sneak preview, look at MESA data and the work of Jim Otvos.
Peter – I’m confused about two of your replies with regards to particle size. Can you clarify?
Above in reply to Lorraine you said: “It’s a size issue, also, and you are correct, the actual molecule of apoB facilitates the endothelial penetration.”
But in reply to Lardlad you said: “Particle size does not matter. The number of LDL particles is the most important feature to understand your risk of atherosclerosis.”
A role for apoB would make sense – if size was critical then you would have more HDL (smaller than LDL) leaking past the endothelium. I’m unconvinced on the concentration gradient aspect. A table in part III states that a good LDL-P is given as <1000 nmol/L and a risky LDL-P is ?1300 nmol/L. I don't see a less than 2-fold concentration difference driving a significant difference in getting past the endothelium.
Peter, as always, thanks taking on this cholesterol thing. I’m a kindergartner here, so if I ask a silly question or somehow missed information that’s already been given, forgive me and point me to the info.
–I was interested to see what you said about veins. I remember reading somewhere that when veins are harvested from one part of the body and placed in service as arteries in another part, the veins can become as atherosclerotic as arteries do. Is that incorrect?
–What causes the excess LDL-P situation in the first place?
–To what extent does the integrity of the artery wall enter into this problem? Are there (I’m guessing nutritional) factors that make the artery wall not up to the task?
–Can high blood pressure — perhaps together with excess numbers of particles — be a factor in the particles entering the artery wall?
High BP makes things worse, probably by causing damage to the intima, which creates even more problems with atheroma and plaques generation. I do not know if higher BP drives the gradient further, but I have seen no data to suggest this.
Sorry. Can’t help myself. Does anyone else see stuffed green olives in the first figure above? Maybe we should stop eating them? . . . .
Quoting from your article,
“Which is the cause – the apoB containing LDL particle OR the immune cells that “overreact” to them and their lipid cargo? … captured by the Robert Burton quote, above. If you don’t know the cause, how can you treat the disease?”
Well, I do understand that you are looking for a “cause” but isn’t the cause in this case the thing that stands out or in other words the abnormality? WBC’s are doing their jobs as they should it’s LDL-P that shouldn’t be hanging around in artery walls. Right?
Secondly,
” In the end, it’s a probabilistic game. The more particles – NOT cholesterol molecules within the particles and not the size of the LDL particles – you have, the more likely the chance a LDL-P is going to ding an endothelial cell, squeeze into the sub-endothelial space and begin the process of atherosclerosis.”
And as you stated a bit above this paragraph, that it’s a gradient driven process. So does this mean that even if you have extremely low LDL-P (as in healthy people) LDL-P molecules STILL can penetrate and reside in arteries ? Because normally arteries have a low concentration of LDL-P molecules, so it’s just that you are merely improving “your chances” of not building up too much LDL-P leading to atherosclerosis, right?
Sorry for the long question!
Less LDL particles, less crossing into the S-E space, less chance of overly robust immune response.
I remember reading that there is a fluid dynamics component to atherosclerosis. Plaques tend to form in areas where there are “eddies” in the blood flow. This would concentrate apoB particles and also provide a physical impetus for penetration of the epithelium (particles flowing directly at the epithelium instead of alongside).
If memory serves, this was also offered as an explanation for the appearance of plaques in arteries closest to the heart, where blood flow is fastest, and not in peripheral arteries or veins.
Excellent article, as usual.
If I may invoke Bernoulli here: In a given dynamical system, the pressure is highest where the flow is slowest (i.e., “eddies”), and where the velocity of the flow is highest, pressure is lowest. (This would mean a bigger effect for arteries, where the total energy present in the fluid would be greater than in the veins.) It begins to make sense to me why high BP is a factor in metabolic syndrome. I suspect that other factors, such as the partial pressures of gases dissolved in the blood, may also be complicating variables.
Thanks for all your work! Maybe I’ve missed it somewhere in the text, but how much does LDL-P and LDL-C correlate? I’ve changed my diet to a ketogenic one, and LDL-C has gone up from 117 to 180.
Coming soon.
Looking forward to this as well. My LDL has gone up significantly since eating low carb. I now feel like I am driving without a seatbelt.
LDL-C or LDL-P? Remember, only the latter matters.
Peter, is there any data on what might constitute an optimum level of LDL-P? There are several studies (Song 1998, Neaton 1992, Waverling-Rijnsburger 1997 & 2003, Razzolini 2008, Cui 2001, Schatz 2001, Schupf 2005, Yang 2008), showing that the all-cause mortality curve vs LDL-C and/or total-C is U-shaped, and there is an optimum LDL-C range somewhere around 100-150 mg/dL. This appears to happen because while CVD risk decreases as LDL-C is lowered, at some point CVD risk is as low as it is going to get, and all-cause mortality increases because of increased risk of cancer, respiratory diseases, accidents, depression, suicide, etc. At low LDL-C levels, these other risks outweigh any further CVD risk reduction. Also, the optimum LDL-C range appears to increase as we get older. It would be interesting if similar data were available for mortality vs LDL-P; do you know of any such studies? Currently, all I’ve seen are statements suggesting that anything below 1000 nmol/L is great, and the lower the better.
I’m able to achieve quite low LDL-P levels by adding Crestor to a very low-carb, high-fat, high saturated fat diet. At a dose of 10 mg of Crestor, my LDL-P is around 300 and LDL-C is 80 mg/dL. With 5 mg Crestor, LDL-P is around 600 nmol/L and LDL-C is 100. Without any Crestor, LDL-P is 1275, and LDL-C is 175. As mentioned above by Dr. Dayspring, this is the opposite of what most people see, my LDL-P is more sensitive to Crestor than LDL-C is. Reverse discordance, perhaps.
So my principal concern is, how low is it safe to drive LDL-P? Are there any studies that might show how much LDL-P suppression is too much?
Excellent questions. I will hopefully have time to get to this when we start to discuss a few the other cholesterol-related topics in the pipeline. I actually JUST had a tutorial on this last month from one of the experts in the fields.
Harry 35:
Do you have CAD, family history? Why do you even take Crestor?
With a particle count of 1275 with an LDL-C of 175 absent CAD or being in a high risk category seems you are in the group of the lucky 25% or so who have lower particle count vs what you would expect given the level of LDL-C.
For obvious reasons, I am going to assume that the conclusion of this series will suggest the best non-pharmaceutical pathway for reducing LDL-P/apoB and thus risk of CV disease is carbohydrate reduction and ideally a ketogenic diet. Hypothetically, if this would still not reduce LDL-P/apoB to an acceptable range, which treatment do you feel the most current research would support? Is this the appropriate time to take a statin? I am guessing this may be anwered in the next post.
Waiting for the next installments with bated breath. (I’ve been low-carbing for about 4 years, now, since diagnosed as prediabetic in early 2008. That, plus exercise, has kept my BGs low and me relatively fit. Still, LDL-P and LDL-C are high (last September, LDL-P, per an NMR test, was 1793; LDL-C was 282). Last December, I had a stent inserted into one of my coronary arteries, and my doctor prescribed 80 mgs Lipitor daily. I am extremely concerned about potential side effects of a dose like that, and haven’t started taking it yet.)
Dr. Dayspring, I wish you were still taking patients (we live in NJ and we called to become new patients of yours but were told no). Would you know of a lab that we can trust to have the HDL kit for the NMR test drawn? Thank you so very much for all you have taught me in your Specialty Health and Lecturepad segments. You are a great teacher, and like Peter you are a great communicator with a wonderful personality!
Thank you Peter for making this understandable. I am looking at an EBT scan with an LAD that has 1 lesion,14.17 volume, and a calcium score of 20.89. Is this atherosclerosis? Is this plaque that burst through the wall into the lumen? Thank you…
Sorry, Maryann. I’m unable to comment on this. Can’t really practice medicine directly like this.
Hi Peter, I completely understand that; I wasn’t seeking any medical advice. I am trying to be a good student and apply the knowledge. I wanted to know if I understand what you are teaching properly.
The coronary calcium scans are interesting, and probably good for an initial assessment. The risk assessment is heavily based on age. So a score of 15 in a 30 year-old is suggestive of high risk, but the same profile in a 60 year-old is not. They should never be used to follow patients or treatment, though. Unfortunately, some doctors feel this is a good idea.
So everyone has this process taking place in their bodies, and what we want to do is minimize it; we can’t stop the particles from penetrating, which is why we want lower particle numbers; we want to stop the cascade that leads to breaking through into the lumen. The calcium score is a measurement of plaque that has broken through, and plaque is atherosclerosis, is that right?
Reducing the number of particles reduces the chance they get into the S-E space. The calcium score only tells you about, obviously, calcium. But it can be misleading. As plaques get absorbed, we can actually see a rise in calcium score, despite a reduction in plaque and risk.
Cholesterol (pron. : kol’ester-oal) is one of the myserty substances of the body. We still know very little about its metabolism. It is excreted by the liver in the bile. It is related to the hormones of sex. It increases in the blood of pregant women, and in some kidney diseases, but its function so far is not well understood.
The Wonderful Story of the Human Body
By A Well-Known Physician
(no date but I am guessing 1930s)
I am still unclear of the influence of diet given that most of the cholesterol we find in LDL and particles is made in our body. Saturated fat ingestion seems to increase LDL levels and particle levels for some. Is this due to an increased burden of cholesterol being delivered to the liver? Thanks.
I will try to address this in future posts.
I am very interested in this as well because I am one of those folks who has seen an increase in LDL from eating low-carb/primal (LDL-P went from 1788 nmol/L to 2112 nmol/L in 8 months while LDL-C had similar values each time, 232 vs. 228 mg/dL). I don’t know what could be the cause(s) other than the diet change but this series has contended that dietary cholesterol isn’t very relevant for those numbers. Previously I wasn’t concerned because I thought I was up-to-date on things with the view that large LDL-P was okay. Maybe I’m not being strict enough on the sugar? I was also recently reading somewhere (I don’t think it was here) that lack of sunlight could create a ‘backlog’ of cholesterol that isn’t being turned into Vitamin D?
Great series, looking forward to suggestions on how to mitigate.
Hopefully we’ll get to the bottom of these issues with more study. Current data aren’t providing enough individual information on some of these issues.
Dr. Attia, First thanks! I am a meteorologist by trade and have always made comparisons between environments. As you wrote about the LDL particles flowing in the serum and how they will become stuck in the endothelium I thought about airflow and how through the right dynamics there is restricted flow due to terrain and how clouds form and persist in locations due to airflow obstruction. An interesting mind twist for me…anyway as you continued with this part:
>>But it seems that not long after an LDL particle gets into the sub-endothelial space and takes up “illegal” residence (i.e., binds to arterial wall proteoglycans), it is subject to oxidative forces and as one would expect an inflammatory response is initiated. The result is full blown mayhem. Immunologic gang warfare breaks out and cells called monocytes and macrophages and mast cells show up to investigate. When they arrive, and find the LDL particle, they do all they can to remove it. In some cases, when there are few LDL particles, the normal immune response is successful. But, it’s a numbers game. When LDL particle invasion becomes incessant, even if the immune cells can remove some of them, it becomes a losing proposition and the actual immune response to the initial problem becomes chronic and maladaptive and expands into the space between the endothelium and the media.<<
I could not help but make a connection to folks crossing the borders…our "illegal aliens" not unlike the LDL particles, you analogies were no doubt the key, but it fits! Not condemning either side in our own illegal alien struggles but do you see the analogy between LDL particles setting up shop, and our own human species "aliens"….something that is part of us, but yet we find invading and so try to remove… interesting! And the fight to remove both is much the same!
Great stuff! Thanks again!
Dave
A doctor friend whom I referred to this web site sent me a link to a cardio-vascular surgeon who supported (i)the re-balancing of Omega 6 and Omega 3, as well as (ii) the position that the low fat-high carb diet was the major factor in the high level of CVD over the last few decades. His position was that high glucose levels in the blood began the inflammation in the arteries, creating the opening for plaque formation. I haven’t the skills to properly comment on his position, but thought it might add to the discussion. https://www.sott.net/articles/show/242516-Heart-Surgeon-Speaks-Out-On-What-Really-Causes-Heart-Disease
Thank you very much. A great intro article to pass to friends.