
Check out more content with expert, Matt Kaeberlein, Ph.D.:
- (Aug 20, 2018) Rapamycin and dogs — man’s best friends? — living longer, healthier lives and turning back the clock on aging and age-related diseases
- (Sep 13, 2021) The biology of aging, rapamycin, and other interventions that target the aging process
- (May 16, 2022) AMA #35: “Anti-Aging” Drugs — NAD+, metformin, & rapamycin
In this “Ask Me Anything” (AMA) episode, Peter is joined by special guest, Dr. Matt Kaeberlein. Together they answer many questions around the field of aging with an emphasis on three specific molecules—NAD, metformin, and rapamycin—and their purported geroprotective qualities. They first discuss aging biomarkers and epigenetic clocks before breaking down the advantages and limitations of the most common experimental models being used today to study aging and pharmacological possibilities for extending lifespan. Next they dive deep into NAD and the much-hyped NAD precursors, nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN). They compare data from NAD precursors to studies on metformin and rapamycin, assessing how they stack up against each other and using the comparison as an opportunity to illustrate how to make sense of new experimental data and make smart decisions about how to approach future research.
We discuss:
- Logic behind comparing NAD precursors to rapamycin and metformin [3:40];
- Aging biomarkers: current state, usefulness, and future promise [7:00];
- Epigenetic clocks: definition, use case, and limitations [14:45];
- Advantages and limitations of studying aging in non-humans and the strengths and weaknesses of different model systems [26:30];
- Aging studies: importance of control lifespans and the problems with reproducibility [34:15];
- Intro to NAD, potential role in aging, relationship to sirtuins, and more [48:15];
- NAD precursors (NR and NMN): current data [1:10:00];
- Human studies with NAD precursors [1:25:45];
- Comparing NAD lifespan data to data from metformin and rapamycin [1:28:30];
- Defining a “clean drug” and a “dirty drug” [1:38:00];
- Reason for the lack of rapamycin studies in humans compared to NAD and metformin [1:41:00];
- Ranking the geroprotective molecules in terms of risk and reward [1:48:00]; and
- More.
Logic behind comparing NAD precursors to rapamycin and metformin [3:40]
The team has been collecting a ton of questions around the science of aging—specifically about three geroprotective molecules: NAD, rapamycin, metformin
*Previous podcasts on related topics:
- Matt Kaeberlein (#10, #175)
- Steve Austad
- Nir Barzilai (#35, #204)
- Joan Mannick
- David Sinclair (#27, #70)
- Lloyd Klickstein
- David Sabatini
The rationale
- Initially, the idea was to exclusively discuss NAD and its precursors
- However, Matt felt strongly that we needed to look at rapamycin and metformin
- The reason being that those three molecules often get talked about together in the field and by people who are following the field as certainly three of the leading candidates for geroprotectors
- There’s a lot of value in a compare and contrast in order to take a look at the current state of the data so that you can really understand what is the evidence for each of these classes of molecules
- Including some of the challenges as we think about moving from the laboratory into the real world, into the clinic in terms of testing them
Aging biomarkers: current state, usefulness, and future promise [7:00]
Are there any biomarkers of aging that we can look at when we look at these molecules?
- When you contrast aging with a field like lipidology, you can see how much more challenging aging is, relatively speaking
- For instance, if your objective is to lower ApoB, because ApoB plays a causative role in atherosclerotic cardiovascular disease, you have the perfect biomarker
- But when it comes to this field of aging, it really is difficult
The two sides of thinking…
- Some people who will argue that we have remarkable biomarkers for aging
- Others argue that we don’t really have any good biomarkers for aging
Where does Matt come down on this?
- One of the things that you have to consider is, what do you want a biomarker to do?
- Note that we’re talking about biomarkers of biological aging
- “What I think you really want is something you can measure that is predictive at either the individual or the population level of future health outcomes, mortality, certainly, but also functional outcomes, disease risk, things like that” says Matt
- On one level we absolutely have biomarkers
- For instance, we can look at two people who are the same chronological age and humans are actually pretty good at estimating who’s in better health
- We’ve evolved to do that so the thinking is that there must be some underlying molecular, biochemical signatures that we can find that are predictive of that
- Trying to find these molecular biomarkers of aging has been going on since the 1980s and it’s still a work in progress
Biomarker candidates
- We do have some candidates now
- There are people in the field who are very optimistic, possibly overly optimistic, about how well those candidates work
- It’s also an interesting time because we’re starting to see commercialization of these so-called aging clocks that are being sold to the general public
- Matt does feel like we are closer than we were 15 or 20 years ago, however, we’re still a ways off from the ultimate goal—having something that you can measure that in a predictive way, at either the individual or the population level, really tells you with any level of precision, what the biological aging trajectory is
The idea of blood-based biomarkers
- Peter says it would be really valuable if we had blood-based biomarkers where you could do interventions for a short period of time
- And if in fact those interventions would, if continued, lead to better lifespan, they would show up
- For example, if you took an individual and you calorie restricted them for three months, took them down to 70% of their weight maintenance caloric intake, you would like to think that there would be some set of biomarkers that would suggest an improvement in their lifespan
What does Matt think about that idea?
- From a pragmatic perspective and a usefulness perspective, that’s exactly what we want
- But it remains a complicated question—it’s one thing to hypothesize that there are going to be molecular biomarkers that reflect biological age, but those are not necessarily going to be the same biomarkers that reflect rate of aging
- A short-term read-out almost has to reflect rate of aging, or even potentially, a reversal of biological aging
- And those may not actually be the same markers for each of those classes
“I certainly believe that there will be signatures of intervention responses that are predictive of efficacy, I’m not sure that it’s going to be the same as the signatures of biological age.” says Matt
Looking back…
- When Matt was first starting in this field 15-20 years ago, if you had asked him then whether caloric restriction is slowing aging or reversing aging, he would’ve answered their slowing aging by decreasing the rate of decline or damage accumulation
- What’s been really exciting over the last 10 years is the observation that at least some of these interventions reverse many of the molecular changes that go along with aging, and in many cases, the functional changes that go along with aging.
Functional biomarkers
- Matt says he’s a big fan of functional biomarkers (i.e., organ function, tissue function)
- It’s harder to do in people than it is in laboratory animals
- Matt feels that functional biomarkers are telling us something fundamental about future health outcomes that you can almost take to the bank
- I.e., if you can make somebody’s heart function better, their brain function better, you got to feel pretty good about that
- If you can make multiple organs and tissues function better with the same intervention, you can probably make a case that you are in fact modulating some underlying biology of aging, as opposed to only the biology of that tissue and organ
“What gives me great confidence that we’re moving in the right direction with a patient, it’s when basically when all of those functional things improve.” —Matt Kaeberlein
- If VO2 max improves, muscle mass improves, strength improves, cardiovascular efficiency improves, phenotypic markers of disease improve (i.e., glucose disposal, insulin signaling, ApoB, lipid markers, inflammatory markers)
- some of those are things you measure in blood,
- some of those are things that you measure non-invasively,
- some of those things are imaging related
- “But until someone comes up with better tools, this is basically how I think about this problem.” says Matt
Epigenetic clocks: definition, use case, and limitations [14:45]
What are epigenetic clocks, how they work, and what they’re aspiring to do?
- The word “epigenetics” actually means a lot
- It can mean anything that is inherited that’s not at the level of your DNA sequence
- But mostly when people talk about epigenetic clocks, what they’re specifically talking about are chemical modifications either to the DNA or to the histones that pack the DNA, and these chemical modifications control gene expressions (such as methylation and acetylation)
- What has been observed in laboratory animals and in humans is that there are changes in these epigenetic marks that happen in a predictable way with age
- There are tens of thousands of these marks that can be measured at any given time in a cell
- You can create algorithms that predict the age related changes in these epigenetic marks with a pretty high degree of accuracy
- For instance, you can sample a subset of specific chemical changes and come up with an algorithm that will predict a person or animal’s chronological age (+/- 5 years)
- It seems to work really well in every organism where people have looked all the way from very early development up into old age, you can create these predictive algorithms
What has emerged from this discovery?
- The idea that has emerged from that is that you can do that at the population level
- If you identify individuals whose chronological age doesn’t match up really, perfectly well with their epigenetic age, then those people may be biologically younger or older than their chronological age
- And that’s where this idea of these epigenetic clocks has come from, is you then at least in principle can predict a person’s biological age, depending on how well they fit the best fit line for this algorithm.
- The evidence in support of that comes mostly from longitudinal studies in humans, where you can create a training set and a test set, and you know what the future outcomes were for some of these people
- You can see a relationship between the people who’s predicted biological epigenetic age is, say younger than their chronological age, and then when you look at them 20 years later they have a lower likelihood of developing specific diseases or potentially of dying
- That’s the case that can be made for these epigenetic clocks, that they are telling you something about future risk
Limitations of epigenetic clocks
- One limitation is that there are about two dozen of them and it’s hard to tell which one works best
- Another limitation, which is more concerning, is that nobody has ever done a definitive experiment which is to actually show in the same individual, or in the same population, that you can actually predict future health outcomes
- Some people will argue that the longitudinal data makes that not necessary, but there are a couple reasons why Matt doesn’t agree with that:
- One is that the environment that we live in as humans has changed dramatically over the last three decades
- And since we know that environment plays a huge role in epigenetic modifications, the epigenetic marks that were most relevant for health outcomes 30 years ago might not be the most relevant today
- The other reason: this is actually a pretty easy experiment to do in mice and it really bothers Matt that nobody has done it
- One is that the environment that we live in as humans has changed dramatically over the last three decades
- Some people will argue that the longitudinal data makes that not necessary, but there are a couple reasons why Matt doesn’t agree with that:
Lack of definitive studies
How many times is someone doing a mouse study that is going to the end of life? Why do we not have the definitive lifespan study for each of these epigenetic clocks?
- People will tell you that the clocks aren’t as good in mice, but it should be doable
- “It’s a black hole in the literature that hasn’t been filled yet.”
The experiment that should be done…
- Take a cohort of mice at say, 20 months, you measure their epigenetic age in blood
- You then do a few interventions that we know should extend lifespan
- You measure their epigenetic age in blood six months later, and then you see at an individual and population level, what the survival is, and you can do end of life pathology
- So if the clocks are working, you should absolutely be able to detect that signature well in advance of end of life
“If somebody did that experiment and it worked, I would be convinced. That would make me really be a believer in the epigenetic clocks, particularly if you could do it at the individual level, but it hasn’t been done yet. So it’s a little bit unclear.” —Matt Kaeberlein
Deciphering the different nomenclature when it comes to the term “epigenetic clocks”
- We use this term broadly and sometimes when a person says epigenetic clock, they mean literally a set of biomarkers that look at methylation patterns on DNA
- Other times when people say epigenetic clock, they mean an algorithm that looks at 15 biomarkers that can include obviously the methylation pattern on DNA, but can include things like vitamin D level, fasting glucose level, traditional biomarkers
Does Matt have a point of view on the difference between these?
- “I would call that more of a general aging clock, punitive aging clock” says Matt
- The punitive aging clocks that incorporate things beyond epigenetics are much more likely to actually work in a useful way in humans
- One reason to believe that is if you look at what people call the nine hallmarks of aging, these famous nine things, molecular processes that seem to contribute to aging, only one of them is epigenetics
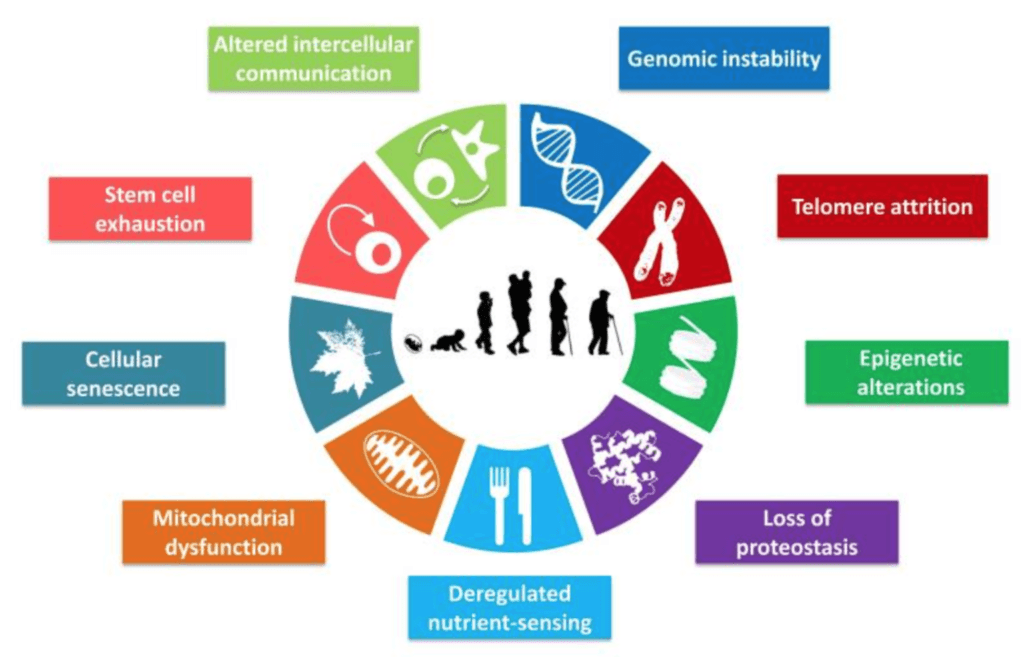
Figure 1. The Nine Hallmarks of Aging. Image credit: López-Otín et al. 2013
- With epigenetic clocks, you run the risk that you’re only informing on a subset of the biological aging processes
- But if you look more broadly, you’re much more likely to get a holistic picture at the whole individual level.
Peter is skeptical of epigenetic clocks
- “I find it hard to believe. I hope I’m wrong, because this would be a really efficient way to do things.” say Peter
- Peter has a hard time believing that there’s going to be an epigenetic signature that will be more valuable than some of the most tried and true phenotype tests:
- Things like VO2 max, zone 2 threshold, grip strength, muscle mass, fat free mass index, and more
- If nothing else, Peter thinks it will be interesting to see how tight that association can be
Matt’s a little more optimistic
- Matt says he’s also skeptical with regards to clocks using methylation specifically, but he’s a little bit more optimistic that you can create the more broad aging clock or aging signature
- Peter asks, “But do you think it can be done out of an existing collection of biomarkers or do you think we’re going to have to go deeper into the proteome and metabolome to find things we don’t even know exist yet?”
- Matt says just given the state of knowledge today that there are a subset of the things that people in the field are thinking about that can actually be extremely predictive at the individual level.
- But all of the things Peter mentioned (VO2 max, zone 2 threshold, grip strength, muscle mass, fat free mass index) and all of the functional outcomes that we know are important for health, there is underlying biology that drives that
- Matt says we’ve got an “incomplete”, but a “pretty good” idea of what a lot of the processes are that are driving that loss of function and degeneration
- The candidates we’ve got are pretty good—and they may not be as precise as you can get if you can do a full functional workup on a person—but they might be good enough to tell you some information about likely efficacy of lifestyle changes or drug interventions or things that people might want to incorporate to potentially maximize their health span
Peter’s point of contention: the changing of the definition of what something means in order to fit a diagnostic test
- Peter is mindful of something he sees a lot when it come to early cancer screening diagnostic companies pitching their products to him
- A common conversation Peter will have goes something like this:
- The company pitching their product with say, “We’ve got a biomarker that is an early detection of cancer”
- Peter says, “Okay, show me the data.”
- Then they say, “Look at this sample set where we predicted so many cancers in patients and we have zero false positives and we have zero false negatives.”
- Peter will look at their data and say, “Well, these are a whole bunch of positives in people that don’t have cancer.”
- They will respond with, “Oh no, no, no, they have early cancer.”
- Peter responds with, “Well, what do you mean by that?“
- They will say, “Well, they have cancer, but it’s only a few thousand cancer cells.”
- Peter will then ask, “But do you know if those people go on to get cancer? Because clinically relevant cancer is about a billion cells.”
- They will say, “No, no, that doesn’t matter. This person has cancer.“
- Peter will then point out, “Well, look, if a person has a thousand cancer cells in their body, we have no idea if that means they’re going to get cancer or if their immune system is going to come along and mop the floor with that cancer. So to tell me, you have no false positives just because you captured those is a little bit like moving the goal post.”
- Peter says he sees a little bit of the same thing going on with biologic age clocks, where there’s pairing an age clock with a supplement or an intervention and there tuning them to each other
- Matt agrees completely and it’s something he’s concerned about as well
- And it’s worth noting that fair number of scientists in the field are concerned about is the commercialization of these aging clocks
Pairing aging clocks with supplements
- Pairing the clocks with supplements is a step further
- “Selling to the general public the idea that, with some level of accuracy, we can measure your biological age and you should take action based on that, is just, frankly, dishonest.” says Matt
- Some people will argue that it’s a necessary evil in the sense that,
- one, it broadens the appeal of the field to the general public
- two, it’s causing people to make healthy lifestyle choices
- I.e., Maybe when you measure your biological age and it tells you’re 10 years older than your chronological age, you start exercising or you eat better
- But it’s still dishonest to claim to people that anyone is able to with any precision measure your biological age, and there are lots and lots of companies doing that
- It becomes a bigger problem when the same companies are then also selling a product that they claim will reverse your biological age – “That’s just snake oil”
- The FDA should step in and do something about it, says Matt
Advantages and limitations of studying aging in non-humans and the strengths and weaknesses of different model systems [26:30]
Peter frames how he thinks about studies of aging
- He starts with a “very MECE bucket” — mutually exclusive, collectively exhaustive bucket
- That “bucket” is the species of interest (humans), and all that is not the species of interest (everything that is not human)
- On the “everything-that-is-not-human list” we’re generally concerning ourselves with eukaryotes and we’ll run the gamut of a billion years—yeast to worms to flies to mammals (mice being the most studied mammal)
- When Peter is looking at a study, he’s looking to see where was the insight gleaned and the closer it gets to the species of interest, potentially, the more interesting it is
- But at the same time, he gets more interested when he sees something that works in yeast and worms and flies and mice… even if you don’t have definitive human data
- He’s more likely to at least speculate in this case that the intervention could work in humans than something that only worked in worms, for example
- But at the same time, he gets more interested when he sees something that works in yeast and worms and flies and mice… even if you don’t have definitive human data
Matt’s response to Peter’s framework above
- Matt would add a third “bucket” to Peter’s two above, that being companion animals (e.g., pet dogs)
- They are outside the lab, evolutionarily maybe a little bit closer to people, but they really pick up that environmental and genetic diversity that we miss in the laboratory
Matt’s take on studying things across billions of years of evolution:
- Matt got his start in the aging field by working in yeast – being mostly interested in genetic modifiers of lifespan
- Then as a postdoc, he started working in C. elegans with exactly the thought that Peter expressed, which is that those things that are shared about the aging process
- Those things that are shared across a billion years of evolution between yeast and nematode worms have a better chance of also being shared in more complicated organisms like mice and potentially people
- That paradigm has held up with the obvious caveat that we still don’t know for sure how much these pathways that seem to affect aging in yeast and worms and flies and mice actually do affect aging in people
- There are some hints that things like insulin signaling, FOXO transcription factors, and mTOR do affect aging in people, but it’s not definitive yet
Each laboratory model has its strengths and weaknesses
- The most common laboratory models that people study are budding yeast, nematode worms, fruit flies and mice
Yeast, worms, and flies studies
- The real strengths of the simple models (worms and yeast, and to some extent flies) are that they age quickly
- This means you can do lifespan experiments in weeks or two months
- You can screen thousands of interventions and find things that affect the aging process just by looking at lifespan
Mouse studies
- When you get up to something like a mouse, they live three to four years in the laboratory
- That’s a much longer term experiment, much more expensive
- And the regulations around working with mice because they’re vertebrae, animals are much more intensive
- There’s a whole underappreciated bureaucratic component to this both in laboratory studies and in why certain clinical trials are done — the bureaucracy drives a lot of both basic and clinical research
- The real advantage of mouse studies is that you can actually look at tissue and organ aging, functionally, molecularly, which means you can really dig a lot deeper into mechanisms, but also convince yourself broadly speaking across the entire body, the intervention that you’re interested in is actually impacting the aging process
- From a pragmatic perspective, the bigger the animal is, the more tissue you can get to actually do experiments
Companion animals studies
- Matt is involved with the Dog Aging Project where they are trying to study aging in pet dogs
- Pet dogs age a little bit slower than mice, making it more challenging
- But the animals of course share the human environment and they get almost all of the same diseases that people do
- So if you find things there, you can have even more confidence that that’s probably relevant for human aging
- But if you’re interested in human aging, you want to do the experiments in humans, but there are lots of reasons why that’s not pragmatic in many cases, one being just the length of time that it takes to really understand, whether your intervention is having an impact on health span and lifespan.
- It’s decades really, to really know the answer for sure, which gets back to what we were talking about before
- Wouldn’t it be nice if we had really predictive biomarkers of biological aging, where we could in a few months feel pretty confident that we’re moving things in the direction?
- obviously that’s where we all want to get to
- “We’ve made progress. We’re not quite there yet, but I’m cautiously optimistic that within a few years we’ll have things that we feel pretty good about.”
Primate studies
- Primate research is very difficult in that they also live quite a long time—a rhesus monkey can live 25, 30 years
- If you look at the calorie restriction experiments, they report that rhesus monkeys can live longer with calorie restriction
- There are other non-human primates like marmosets, which are shorter lived.
- There are people studying aging in marmosets but one of the real challenges with something like a marmoset is they haven’t been used extensively in any area of biomedical research so we actually don’t know a lot about the normal aging of marmosets
- From talking to some of the investigators involved in aging studies in marmosets, Matt says that the researchers have actually been a little bit surprised because the lifespan for marmosets actually seemed to be a little bit short and their animals are living longer than they expected them to live
- With marmosets, there’s just not this foundation of data about what normal aging looks like, which makes it really hard to design well-powered, well-controlled experiments to study the effect of an intervention on aging in those kinds of non-traditional animal models.
Aging studies: importance of control lifespans and the problems with reproducibility [34:15]
Understanding the lifespan and the control group
- Speaking about mouse studies (most common with aging studies)
- Matt says that what actually crystallized in his mind was seeing the literature in C. elegans’ experiments over the years and how variable the control lifespan was for what should be the same genetic background, the same temperature, the same media conditions
- Sometimes you would see a 50% (sometimes even a 100%) difference in the lifespan of the control strains across studies
- Many of the lifespan extending interventions—both genetic knockdowns or drugs—were reported in experiments where the controls were short lived were not reproducible in subsequent studies
- It’s possible that the irreproducibility that we see in the general science literature might come from experiments where control strains were short lived for reasons we don’t understand
- Therefore, it could be that the “lifespan-extending” intervention really just brought the controls back to where they should have been
- In many cases, reviewers are hesitant to go to people after a three year experiment and say, “Your controls are really short lived. You need to go back and do that experiment again.”
- That’s the right thing to do in Matt’s view, but it’s hard to ask people to do that
- Mouse aging experiments are rarely attempted to be replicated—so if somebody reports a genetic intervention extends lifespan by 30% in mice, that doesn’t get tested again for many years, if ever.
- We have several cases now where when researchers actually do go back and try to replicate these things that they’re unable to replicate them
- And many times this can be traced back to the absolute lifespan of the controls
Looking deeper into this issue with the lifespan of the control groups…
- Matt looked into this issue and found there was a pretty big variation in the control lifespans by just looking at the mean lifespans that are reported in the papers
- In the data, Matt extracted an apples to apples comparison—meaning he looked at the male mice all in the same strain background
- He analyzed studies that used four interventions: nicotinamide riboside, metformin, alpha-ketoglutarate, and rapamycin
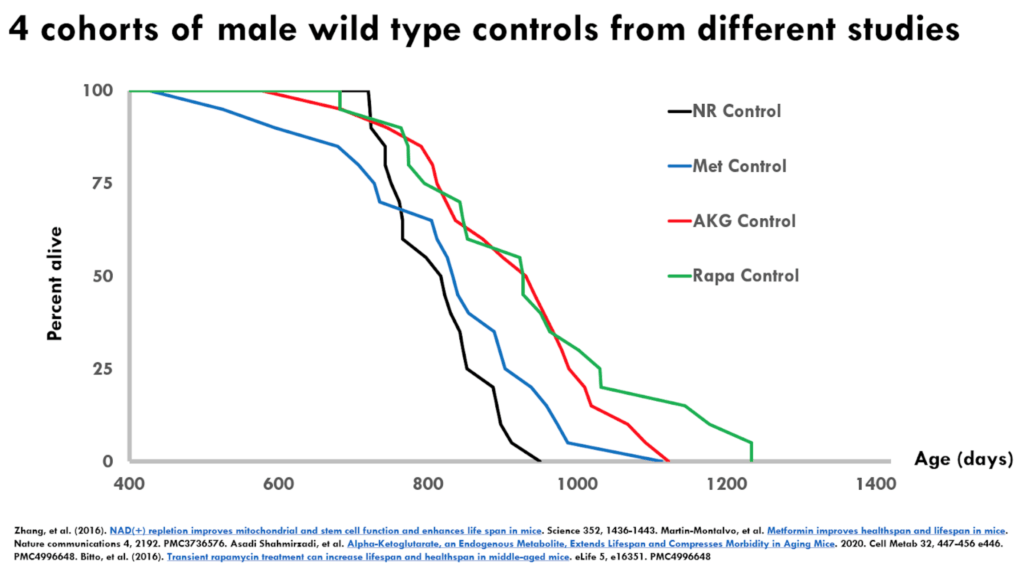
Figure 2. 4 controls with wild type controls from different studies.
This is a Kaplan-Meier survival curve—this is by definition, a monotonically reducing function because it is a cumulative incidence of death
- You can see on the X axis that it is showing you at 400 days is when they’re starting to keep track where 100% of the animals are alive
- With each passing day, there’s basically two options: That number stays the same or that number goes down, but it can never go up
- You can see these numbers all come down and eventually they cross the X axis at which point all of the animals in the respective control groups are dead
- You have four different lines representing four different studies
- There are all the male C57 black 6 mice
- The lines represent the wild type untreated controls from each of these studies
- This is just showing days from birth and the percent alive
Important point to understand: If everything worked perfectly, these curves should be identical because in principle they are genetically identical animals treated the same way all in pathogen-free conditions
- But what you see is that the curves are not on top of each other and in fact, they are pretty well spaced apart
- Note that something that is shifted to the right is typically thought to be extending lifespan so people will claim that their treatment extends lifespan if the survival curve is shifted to the right
- If we just were to look at these four curves, there would be statistically significant differences and very low P-values between the black and the blue being the shorter lived, and the red and the green being the longer lived, even though these should all be in principle the same
- Another point: Even though the blue and the red would share the same maximal lifespan, Peter says “I have to think that the blue and the red would differ statistically in a median lifespan shift. In other words, you could easily look at this and think the black is the control, the blue, the red and the green are three different lifespan extending strategies.”
- Not only that, but red and the green would be considered very large lifespan extensions for mice
Important point about the maximum and median lifespan extension:
- The stat that almost always gets presented is the median lifespan extension
- Another thing is that Matt doesn’t like the fact that the stat presented is always “percent” lifespan extension because it’s very susceptible to short lived controls
- When the denominator is smaller, the percent effect is going to be greater
- If you have a really, really short lived control, you can get a 30-40% increase in lifespan, that is pretty much meaningless because your control was so short lived
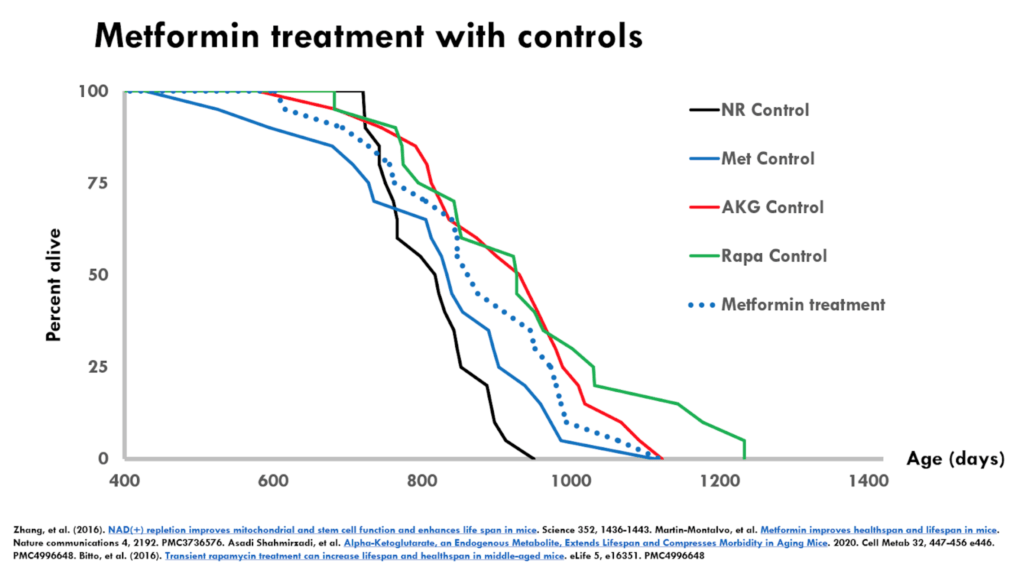
Figure 3. Metformin treatment with controls.
- In the figure above, we’re looking at the metformin treatment superimposed on all four controls–including the metformin control
- The dotted blue line is the metformin treatment and the control for this experiment is the solid blue line
- When you compare just the dotted blue line with the solid blue line, you will come to the conclusion that metformin increased median lifespan
- If you look at the average of the controls, you would suspect that metformin is no different than the average of the control
- Does that mean that metformin didn’t extend lifespan? ⇒ “No, I wouldn’t say that. But it also makes me concerned that this is not going to be a robust and reproducible effect because the treatment group in this case was actually shorter lived than some of the controls from other studies.”
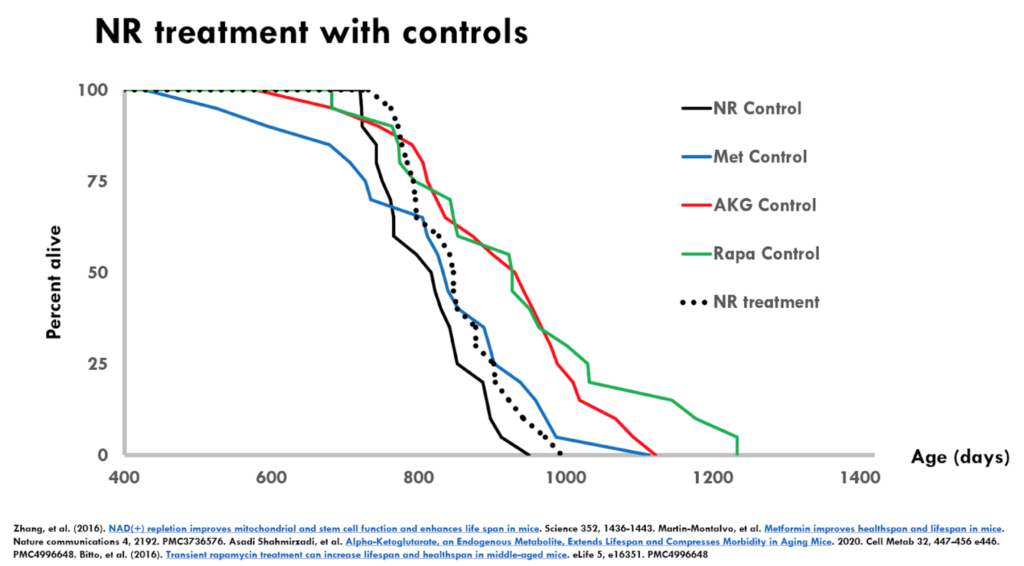
Figure 4. NR treatment with controls.
- The figure above is the nicotinamide riboside (NR) experiment
- The dotted black is the NR treatment group and the solid black is the control from that same experiment
- The results are almost an identical interpretation to metformin
- In this case, the treated group is even shorter lived than the metformin treated group
- It raises the same sorts of questions about the robustness and the reproducibility of this result.
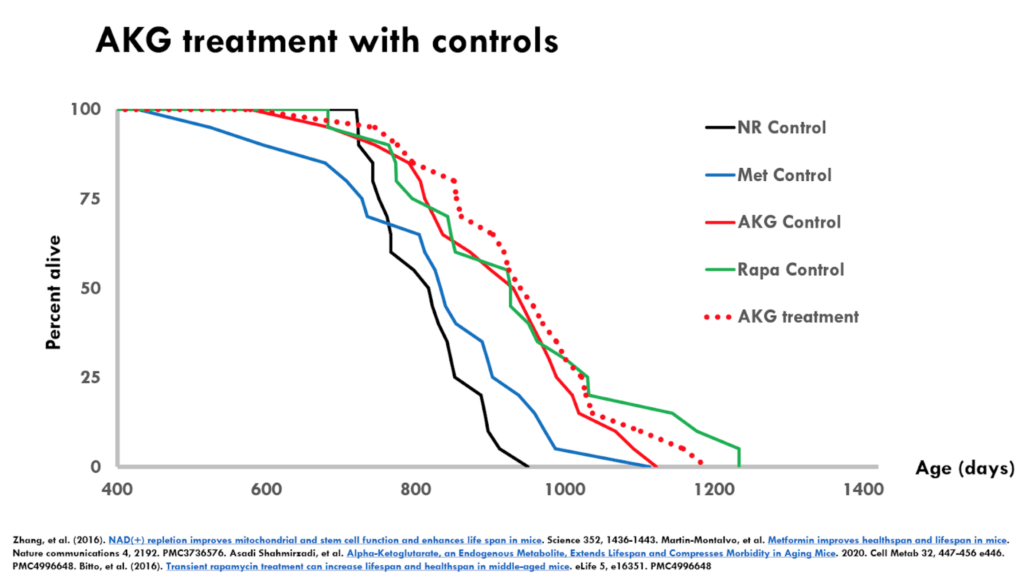
Figure 5. AKG treatment with controls.
- The figure above is the alpha-ketoglutarate experiment
- It’s an interesting example where the controls were pretty long lived
- The treatment has a even smaller effect relative to the experiment-matched controls than either metformin or NR
- But because the controls were so long lived, you might feel a little bit better about alpha-ketoglutarate in terms of effects on lifespan
- And certainly if you compare the effect of alpha-ketoglutarate to say the metformin control, there’s a huge effect there
- If this is the only data that we’ve got, there might be something there with alpha-ketoglutarate but we need additional data
Another point to make: We’re looking only at lifespan data
- Many of these papers have other measures that can strengthen the hypothesis that they are affecting the aging process
“Personally, I’m not a big believer in studies that claim that an intervention had major effects on health span if it didn’t do anything to lifespan. But I think you do want to take into account, this is only looking at lifespan and not looking at other functional measures of aging.” —Matt Kaeberlein
Matt’s 2016 rapamycin paper [44:15]
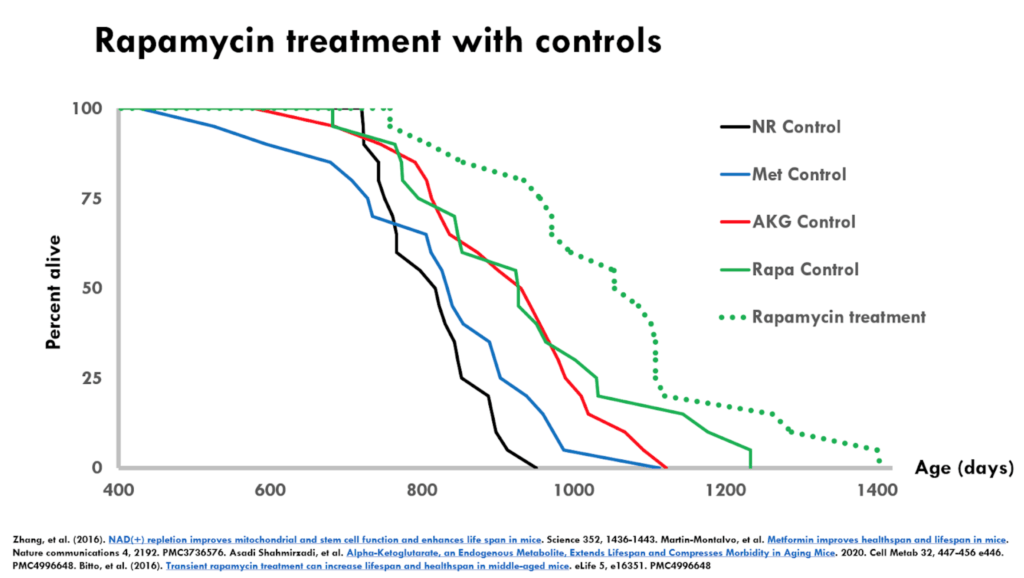
Figure 6. Rapamycin treatment with controls.
- This is a dose of rapamycin that had a large effect on survival
- The dotted green line (treatment group) is far to the right of the solid green (control group)
- This as a solid result because of the magnitude of the effect over long lived controls
- It’s even more interesting because rapamycin has been shown by many other labs to extend lifespan
- One of the interesting things about this particular comparison is the two interventions that supposedly extended lifespan with short lived controls, failed to show lifespan extension when they were independently tested by the interventions testing program (ITP with NR, ITP with metformin)
⇒ Peter spoke with Rich Miller about the ITP studies of rapamycin
More about the ITP
- The ITP uses a strain of mice (HET3)that are not heterozygous at all loci — meaning they are not inbred to the point of being identical
- They’re a cross of four different genetic backgrounds—they’re genetically heterogeneous, certainly compared to any one inbred strain background.
- The ITP carries out three parallel experiments simultaneously in three separate labs
- The ITP studies have repeatedly shown rapamycin extends life
Why Matt has more confidence in ITP studies
- One is the genetic background being heterogeneous
- Second is the built-in triplicate replication that you mentioned that all of the studies are done at three independent sites
- Generally the pool data matches the data from each site
- Third is the ITP investigators have no vested interest in the outcome of the experiment
- “when you have a vested interest in the outcome of a study turning out a certain way, you may not change the data but I think you tend to interpret it in a way that favors the outcome that you want”
- we see this in the all literature, especially in the aging field
- Often those same authors have significant interests in that model becoming accepted among the broader community
Intro to NAD, potential role in aging, relationship to sirtuins, and more [48:15]
NAD 101
- NAD is a molecule that is present in all cells, central to more than 500 different metabolic reactions. Plays a key role in carbon metabolism, redox homeostasis.
- It’s essential for life—if we are deficient in our ability to synthesize or obtain NAD cells can’t survive.
- But also in some ways, very difficult to study because it impacts so many different aspects of cellular function.
- “In fact, it’s actually hard for me to think of a cellular function that isn’t affected by NAD because it’s centrally involved in metabolism and energy production and redox homeostasis”
- Difficult to explain this stuff, if a person hasn’t taken organic chemistry and doesn’t understand the purpose of sharing electrons
Example of purpose of sharing electrons:
- the electron transport chain: we turn those chemical bonds into an electron gradient, and then back into chemical energy in the form of ATP
- A lot of this has to do with shuffling electrons—somebody needs to accept and pass those things along in NAD and NADH do so much of that work in the mitochondria
Relationship to sirtuins
What are sirtuins? ⇒ Sirtuins are a family of enzymes that are highly conserved
- Matt started studying sirtuins when he was a graduate student working in yeast
- He was studying a protein called Sir2, which is actually where sirtuins get their name from
- At that time, they didn’t know what the biochemical activity of Sir2 or other sirtuins was
- They were studying it in the context of aging and part of my PhD thesis was work showing that if we overexpressed Sir2, we could extend lifespan in yeast
About the same time, a postdoc in Matt’s lab, was working on trying to understand the biochemical activity of Sir2 and the mammalian homolog, which is called SIRT1
- The postdoc discovered that Sir2 was what we call a histone deacetylase, meaning it takes acetyl groups off of histones, which are the proteins that pack DNA
- Histone acetylation is one of the types of epigenetic marks that regulates gene expression
- What the postdoc found was that the Sir2 protein could remove those acetyl groups from histones, which opens up the gene expression of the genes where those histones are bound
- The thing that was most interesting about this was that it was a brand new type of histone deacetylase, in terms of its biochemical mechanism in that it used NAD.
- So it defined a new class of chemical reaction called NAD-dependent histone deacetylation
The way this works: unlike many of the metabolic reactions that NAD is involved in, where NAD gets converted to NADH or vice versa, sirtuins actually consume NAD
- It’s not an interconversion of NAD to NADH based on its redox state…NAD actually gets broken down into ADP ribose and Nicotinamide as the acetyl group gets taken off
- So that means NAD is actually a substrate of the sirtuins that gets consumed in this deacetylation reaction
That’s very different from what’s happening in most of the NAD NADH interaction where it’s not being consumed, it’s being shuffled in form
- There’s probably 500 or 600 reactions or NAD is converted to NADH or vice versa
- There are only a few types of reactions where NAD is actually consumed. And sirtuins are one of the classes of enzymes that do that
- this is a really important point to appreciate — these are fundamentally different ways of affecting NAD biology. You can interconvert it from NAD to NADH or you can actually break down the NAD or synthesize NAD as needed with other types of chemical reactions
- It’s still a little bit of an open question about how important this break down of NAD is by sirtuins versus overall NAD homeostasis
- There are other enzymes that break down NAD that many people think are probably more important than sirtuins are
Do sirtuins promote longevity?
- The key point from the model that is still widely considered in the aging field is this idea that sirtuins promote longevity
- If sirtuins do promote longevity outside of yeast, is that activity of sirtuins, the longevity promoting activity dependent on NAD?
- If so, maybe you can regulate longevity through sirtuins by manipulating NAD levels
- That’s the central hypothesis to much of the idea that’s out there about the importance of NAD for aging
“There’s a lot to unpack there, and I’m not a big believer that that hypothesis is strongly supported, but that’s the big idea that NAD regulates sirtuin activity, and sirtuin activity regulates longevity.” —Matt Kaeberlein
2000 paper, Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae (Lenny, Su-Ju Lin)
- Paper was looking at the ability of caloric restriction to extend lifespan in yeast
- NOTE:
- The way that caloric restriction was studied in yeast is by reducing the amount of glucose that’s in the media
- that’s important because this is unique to yeast—human cells don’t do the same thing—in yeast when there’s lots of glucose around, they primarily ferment that glucose to ethanol
- This is great, but it’s not exactly the same metabolism as most other eukaryotes carry out
- And when you restrict glucose, they actually shift over to more of a mitochondrial form of metabolism, respiration, which is what our cells typically favor.
- So that changes the importance of NAD when you switch from a fermentative metabolism to a respiratory metabolism
- What they found:
- if you made MUTATIONS in enzymes involved in NAD biosynthesis, that you could block the lifespan extension from caloric restriction
- the same thing was true if you deleted Sir2, that caloric restriction no longer extended lifespan, if you deleted this Sir2 gene, which again is an NAD-dependent histone deacetylase
- that led to the model that caloric restriction is acting through Sir2 by activating Sir2, by increasing NAD levels
- That model, Matt thinks, was supported by the work that I alluded to earlier, that I did, where we showed that if you overexpressed Sir2, you could extend lifespan
- The pieces were there, there are obviously alternative explanations, but the pieces are there
- We knew that overexpressing Sir2 was sufficient to extend lifespan.
- Peter asks: “But Matt, was it NAD-limited?”
- That’s a harder question to answer
- We knew that Sir2 was an NAD-dependent enzyme
- Peter follows up: “But you’d have to make the case that you’re NAD-limited if increasing NAD is the answer to that question outside of caloric restriction. Wouldn’t you?”
- This gets to how much evidence for a given model do you need?
- What was shown at the time was that if you depleted NAD genetically, you could block the lifespan extension from caloric restriction
- This happens all the time in the literature: There’s the necessary part. And there’s the sufficient part. And oftentimes not both pieces are present in the same paper
- Matt says, “what I would say is in that paper, the data all supported the model that caloric restriction increased NAD activated Sir2, activated Sir2 extends lifespan”
- “It’s a very simple model. It’s elegant. Everybody likes simple elegant models. All the data I just told you about, supported that.”
- Something missing from the paper:
- There was one piece of data that many people in the lab at the time knew about, but that didn’t happen to make it into the paper, which is that the strain background where I overexpressed SIR-2, it’s called W303 just for historical perspective, did not respond to caloric restrictions
- So you could overexpress SIR-2 and extend lifespan, but if you restricted glucose, you did not extend lifespan in that genetic background
- The genetic background where all of these other experiments were done in, which is called PSY316, had the opposite phenotype
- You could overexpress SIR-2, it didn’t do anything to lifespan, but caloric restriction extended lifespan
- So that was a complicating piece of data that was difficult to explain, “but it made me a little bit worried that there was something about this model that we didn’t quite understand”
Picking back up the yeast-aging story:
- Matt left the lab and spent about a year and a half in biotech, and then came back to academia and joined Stan Fields’ lab at the University of Washington
- Matt teamed up with Brian Kennedy to kind of pick back up the yeast aging story
- They wanted to try to find stuff other than SIR-2, because really everybody was focused on SIR-2 at that point
- They started working in a third strain background, which was the genetic background called the deletion collection—a set of several thousand single gene deletions in yeast
- Their real interest was in doing an unbiased genetic screen for all the single gene deletions that were viable and looking for things that extended lifespan
- As a start, they overexpressed SIR-2 in that background and it extended lifespan
- Next they calorically restricted and it also independently extended lifespan
- So the two interventions worked independently, so they both extended lifespan in this third genetic background
- This allowed Matt to do an experiment that couldn’t have been done before, which is to say, well, what happens if we combine those two?
- If they’re in the same pathway, the prediction is, it shouldn’t do much
- But what we found was a clear additive increase in lifespan
- That got them thinking that maybe the model wasn’t quite as straight forward as they thought
- At this time, the definitive experiment would be if we could get rid of SIR-2 and show that we could extend lifespan with caloric restriction. That kind of disproves the simplest version of that model
- The problem with a SIR-2 single mutant is it’s very short lived—the mechanism has to do with a hyper recombination in the RDNA
- But they could make a second mutation that blocked that thing that made SIR-2 mutants sick so they then had a wild type lifespan (this is a double mutant now called SIR-2 FAB-1)
- This mutant has a wild type lifespan, but it has no SIR-2
- They calorically restricted that mutant and got a massive increase in lifespan, even bigger than you see in wild type cells
Matt published a paper that basically said that lifespan extension from caloric restriction was independent of SIR-2
Matt says…
- “I tend to be pretty skeptical of my own models, and I think you could still come up with an alternative hypothesis that maybe under some conditions, caloric restriction activates SIR-2 and other conditions it doesn’t…
- …I’m not even saying that’s not true, but nobody has actually done any experiments to support that idea.”
- (Peter adds that the best case scenario for sirtuins is that “at most it’s sufficient, but not necessary.”)
Matt continues:
- As people have gone on and tried to test this in other organisms, there are hints that you can activate sirtuins and worms and flies and increase lifespan
- There’s other data that hasn’t been able to reproduce that
- So it’s still a little bit unclear to what extent you can robustly increase longevity outside of yeast by activating sirtuins
- “I’m not saying that sirtuins aren’t important for longevity. There’s just not a lot of really, really strong data to demonstrate that, even in mice.”
- There’s a lot of cases where data couldn’t be reproduced
“It’s really hard to know, in my view, how important sirtuins are as longevity factors…If we accept that, then it’s difficult to know [the importance of] activation of sirtuins by NAD as a longevity mechanism.” —Matt Kaeberlein
NAD and mitochondrial function
Mitochondrial dysfunction is indeed one of those hallmarks of aging
- If compromised or impaired redox signaling goes part and parcel with compromised metabolic function, there’s a “hand waving” way to say that more NAD has to be better
- Matt agrees that “It’s a very hand-wavy argument, but it does make sense”
- When you have mitochondrial damage (this could also happen in glycolysis, central carbon metabolism) that feeds some of the substrates for mitochondrial respiration, you go from glycolysis into the TCA cycle, which is in the mitochondria, to drive the electron transport chain
- These perturbations in those central metabolic reactions, which all use NAD, you could in a hand wavy way, think that would affect NAD homeostasis and that by replenishing NAD that could be beneficial
⇒ Specific example:
- There are a variety of pathological conditions where you have mitochondrial dysfunction that impairs electron transport chain function
- What tends to happen is your NAD all gets consumed through glycolysis
- We hear about the mitochondria being the powerhouses of the cell, and really what we mean by that is mitochondria produce ATP, which is the chemical energy currency of the cell.
- It turns out glycolysis is actually much faster at making ATP, if you just churn carbon through glycolysis, without going into the mitochondria
- It turns out when your electron transport chain is impaired, you can make plenty of ATP through glycolysis
- What happens though is your NAD gets all converted to NADH because the way you take your NADH, the reduced form of NAD, and bring it back to NAD is through the electron transport chain
- If you can’t do that, you get a lot of NADH built up
- The way to solve the problem of your electron transport chain not working is through lactate fermentation
- That’s this idea that if you’re exercising really hard, some of your cells will produce lactate—It’s to restore NAD
- Under certain cases where your electron transport chain is impaired and NAD homeostasis gets pushed towards NADH, and you need that NAD to keep glycolysis going, that restoring NADH from NAD homeostasis could be actually quite beneficial
- Experiments in mice have worked where you have mitochondrial defects and restoring NADH homeostasis through the use of these NAD precursors
- So that you can actually have quite beneficial effects in the context of these mitochondrial dysfunction syndromes
Does NAD help an aging individual via DNA repair?
- PARPs (poly(ADP-ribose) polymerases) using NAD for DNA repair
- We know that DNA damage increases with age, therefore, presumably the demand on DNA repair increases with age (NAD being an important part of that configuration)
What’s the evidence for this seemingly very plausible hypothesis that: Giving NAD to an aging individual, could improve their longevity?
- There’s maybe another layer there, which is that NAD or NAD homeostasis will decline with age and that by restoring that you could improve lifespan, improve health span outcomes
- But NAD is not something that can be taken orally, but it certainly can be given intravenously
What do we know about intravenous NAD? Does it solve this problem?
- Matt is not aware of any data that convincingly shows that intravenous NAD in healthy people has any benefit (or any detrimental effect)
- And there are questions around whether or not intravenous NAD itself actually has an effect on NAD homeostasis/NAD levels in cells
Outside of some cells in the brain, Peter doesn’t think there are any cells in the periphery that even have an NAD transporter. He asks Matt, “Can we get intravenous NAD into a cell?”
- Matt says his understanding is that that’s unclear, there’s not much evidence to support that
- But it’s certainly possible that at some point, somebody will discover an NAD transporter that can do that
- NAD is not able, by itself, to cross a lipid bilayer, get across the cellular membrane and so you need a transporter to get it into cells
NAD precursors:
- Having said that NAD and the intermediates in the NAD biosynthesis and breakdown pathways are highly interconvertible
- This is something that is emerging, that our cells are really quite able to take up precursors of NAD in that NAD pathway.
- There could be a salvage pattern here where if you give somebody intravenous NAD, it’s possible that they’re turning it into NMN or NR… and then right back to NAD in the cell, if the NMN or NR gets transported into the cell
- People are studying NAD precursors (nicotinamide riboside and nicotinamide mononucleotide) primarily in the context of aging
- There are still real questions around the efficacy with which those molecules are taken up through the gut once they get into circulation taken into cells
- But there are other NAD metabolites like nicotinamide, which can be interconverted
- What seems to be the case is that even when you treat with, say an NAD cursor, it may not get into circulation as those molecules, but they can be interconverted in circulation or in cells
- The real questions are:
- 1 – Do those molecules have a big effect on NAD homeostasis and NAD levels in cells and tissues?
- 2 – Do we see functional improvements in those cells and tissues that then you would predict would turn into improved health span, potentially increased lifespan in laboratory animals?
NAD precursors (NR and NMN): current data [1:10:00]
Figure 7. Source: nmn.com
What’s with all the hype?
- Two molecules, NR and NMN
- These are orally bioavailable
- They are safe from an acute toxicity standpoint, there’s anything that one needs to be concerned with here
- These are over the counter supplements
- For as much hype as there is around this, we really don’t have a lot of great data to point to that these things are improving lifespan
What does Matt make of this hype in the face of a lack of clear data?
- This is the crux of the issue
- When evaluating different interventions that potentially affect the biology of aging, Matt asks himself: What’s the best data that these things actually have a strong impact?
- And in this particular case, when referring to normative aging…
- we don’t want to be working in a short-lived background that has some specific pathology
- And in this scenario, Matt is not confident that NR or NMN can increase lifespan in wild type mice
So what is the most compelling evidence?
- Well, the only time one of these molecules has gone to the ITP was nicotinamide riboside, and it failed to extend lifespan
Study by Zhang et al., 2016:
- This study reported that late life administration of nicotinamide riboside in C57 black 6 mice had a small, but statistically significant effect on survival
- As was noted in Figure 4 above, the controls in that experiment were shortlived and the experiments where the controls are shortlived, if the effect of the intervention small, that’s often not reproducible
- And in this case, when the ITP attempted to test nicotinamide riboside, they saw no effect on lifespan or any other real phenotype
- One limitation from the ITP is they did not actually show that the nicotinamide riboside in those animals had an effect on NAD levels or NAD homeostasis.
- What they did do was look at a separate cohort of animals treated with nicotinamide riboside, but even there, while there were some changes in NAD metabolites, it’s not clear to me that they actually had the anticipated effect on NAD levels
- it’s important to say that it’s not completely clear that treating mice with nicotinamide riboside actually has the anticipated effect on NAD levels, at least in a variety tissues
- Sp it’s been a real challenge to interpret the literature, “because for reasons I don’t understand, it hasn’t been clearly reproducible that treatment with nicotinamide riboside actually increases NAD levels in mice”
Peter adds: The question that always goes with that for me is: Where should one see NAD levels go up? Should it go up in the plasma? Should it go up in the PBMC? Should it go up in the tissue?
- This creates a bit of a conundrum which is: when this thing doesn’t work, which it clearly doesn’t work in most experiments to extend lifespan, we don’t actually know if the hypothesis has been tested adequately
For instance, there’s lots of data that look at both temperature and moisture stability of these molecules and Peter is “surprised at how poor it is”
- Matt says, “First of all, it’s not clear to me exactly what the relative stability of say nicotinamide riboside and nicotinamide mononucleotide tested side by side under the same conditions is”
- There’s an additional question here: Even if it’s a hundred percent stable, how is it getting into the animals? And is it effective at increasing NAD levels?
To the question of where would you expect to see changes in NAD:
- Matt would argue if you’re expecting this to be a broad spectrum drug that affects the biology of aging, you would expect to see changes in the tissues where NAD homeostasis is most perturbed or most important for the aging process
- In Matt’s lab, they’ve tried both nicotinamide riboside and nicotinamide mononucleotide in the context of a mitochondrial disease model and they never saw effects from either one in terms of disease progression
- What he DID see with NMN was a change in NAD levels in liver, but not anywhere else
- Matt says to “take this with a grain of salt” because this was not well controlled
- What he did see was what looked like an impact on proteins acetylation in mitochondria in liver
- To give a little bit of background:
- The primary mitochondrial deacetylase is SIRT3, another one of these sirtuin enzymes
- Mitochondrial acetylation is often used as a proxy for NAD homeostasis in the liver, because it seems to be the case that SIRT3 is very responsive to NAD levels
- So if you increase NAD you can increase SIRT3 activity and decrease protein acetylation in the mitochondria
- What Matt saw was a change in NAD levels and a change in protein deacetylation in liver mitochondria
- That made him believe that they probably affected NAD homeostasis in the liver, but not in the brain, for example
- This has made him wonder if many times these NAD precursors are preferentially impacting the liver, and then just not actually having an impact in other tissues (just speculation)
- This is partly why a lot of times those kinds of results don’t ever make it into the literature, you can’t spend years and years studying stuff that doesn’t work
- Matt’s theory is that a lot of people have probably tried to get nicotinamide mononucleotide and nicotinamide riboside to work, and when it didn’t, they gave up
- This is a challenge with some of these things that get a reputation in the literature for having all sorts effects
- You don’t actually see all of the times that people tried it and it didn’t work because there’s a huge bias against negative publication, unfortunately
Where NAD precursors could be effective:
- Matt believes there are certain pathological conditions where NAD homeostasis is perturbed so much that it creates a major problem
- And the data there looked pretty good
- There are studies by Vilhelm Bohr:
- He’s working in these mouse models of high levels of DNA damage where you get hyperactivation of PARP and depletion of NAD
- In those mice, nicotinamide riboside seems to work pretty well
- There is a significant improvement in function and survival
- There’s also a mitochondrial disease model where NMN partially rescued the survival deficit
How Matt thinks about NAD:
- If you think about how important NAD is in a cell—it probably has 500,000 different targets that it interacts with—cells have to buffer that really well
- Cells can’t tolerate NAD dyshomeostasis to any significant extent, or there’s going to be big problems: When you get massive NAD dyshomeostasis, there are big problems
- If NAD levels really decline during aging, do they decline enough that it creates problems, that it moves the cells out of that range where they’re buffered?
- That’s an unknown question
- Secondarily, when you give these precursors, are those going to be enough to move the cells out of the range where they’re buffered?
- Because NAD can be interconverted to all sorts of stuff (we haven’t even talked about NADP and NADPH)
- So it’s not clear to Matt that the dyshomeostasis is enough that these precursors can have an effect
- They may very well be bioavailable, it’s just really hard to measure because you’re working in that range of buffered homeostasis, and it’s only when you get outside that range where you have severe pathology, where you can actually see some of the benefits of these NAD precursors
Where should the research go from here?
Peter would like to see:
- 1 – the doses are going to have to be a lot higher than they’re being proposed
- the recommendations for NR and NMN dosing, we’re talking about something in the neighborhood of 500 milligrams daily
- why not make that two grams a day or something?
- 2 – stability issue figured out
- We want to be clear on this stability issue
- Does this need to be a crystalline structure? What needs to be done to maintain the stability of this molecule
- 3 – more “definitive” studies
- given how much commercial interest is in this space, it amazes me how many of the “definitive” experiments haven’t been done
- Why aren’t there more studies?
- 1 – Because they don’t have to as this is not a regulated pharmaceutical
- 2 – They don’t really want to because they might not get the answer that they’re looking for
Matt makes a point about going to high with the dose:
- NAD has to be buffered really well in the cell and if you go out that range on the low end, there are real problems that happen
- But also, there might be real problems that happen if you go out of that range on the high end.
- He doesn’t know how much NAD precursor you would have to take to bypass that buffering that’s built in
- But he would actually be really worried about given all the reactions that NAD’s involved in if you push NAD out of that homeostatic range on the high end
- It’s probably going to be really hard to get enough NAD precursor into the system to push NAD out of that buffered range, but if you do it, things might go off the rails pretty quickly
Cancer risk with NR?
In a previous podcast with Inigo San Millan,
- Inigo talked about a very, very small experiment he did: four mice in the control and four mice in the treatment
- Mice had a very aggressive cancer — it was a triple negative breast cancer in the eight animals
- Turned out that supplemental NR accelerated cancer growth.
- To be clear, accelerating cancer in a cancer prone model is not the same as causing cancer
- Peter isn’t saying that these supplements cause cancer, no evidence to suggest that
- It might be like growth hormone — Peter doesn’t find the evidence remotely compelling that growth hormone causes cancer, but it would be very difficult to justify anybody with cancer should be taking growth hormone or any anabolic hormone for that matter
- Peter wonders: I wonder how we’re going to get to questions like that?
- Matt says it’s a tough thing to tease apart
- You can certainly do kinds of experiments in mice
- Mice are challenging obviously for cancer research in particular — everybody recognizes that many things that cause cancer or prevent cancer in a mouse, don’t do the same thing in people
- It’s going to be really hard to, from a mechanistic perspective tie the kinds of effects like you were just describing to one thing, because of all the metabolic reactions that NAD is involved in and how centrally tied glycolysis is to cell division and ATP production and the Warburg effect
- it would not be surprising if pushing NAD too far towards the high end would impact central carbon metabolism and glycolysis in a way that if the cells are all already primed to go down that path that you might accelerate that, or you might enhance that
Human studies with NAD precursors [1:25:45]
Are there any really impressive studies done in humans on these molecule?
One that comes to mind is the ALS trial from three years ago
- A small study looking at nicotinamide riboside alongside a sirtuin activator
- The study did show a delay in the progression of ALS
- Matt says there are going to be some disease conditions where NAD homeostasis is significantly perturbed that leads to the disease
- There’s a pretty good therapeutic opportunity if you can restore NAD homeostasis to have an impact in those disease conditions
- The ALS study may be an example
Science paper: Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women
- Very small study of about 25 women
- Treated with NMN or a placebo
- Despite what the authors reported, Matt is not convinced by the data in the paper that insulin sensitivity was actually improved
- Also, the groups were assorted between placebo and treatment group in a way where there was a significant difference in hepatic lipid levels between the placebo and the treatment group
- And given that that’s one thing that these precursors have been suggested to do is affect metabolism in the liver, fat metabolism, in particular, but that complicates the outcomes in that study
- So even if you accept the outcomes, that complicates the outcomes because the randomization was not appropriate in this sense
Comparing NAD lifespan data to data from metformin and rapamycin [1:28:30]
Metformin and rapamycin
What are the histories of these two molecules and why are they being proposed as gero-protective molecules in the first place?
- The first studies of metformin for lifespan actually sprang out of earlier studies that were done in the ’80s by a Russian scientist named Oleg Anisimov, where he was studying phenformin—a precursor of metformin
- He had this hypothesis about phenformin and biguanides affecting fatty acid metabolism, in some way, that being relevant for aging
- He did some lifespan experiments in a short-lived cancer-prone mouse strain, where they saw, first of all, that phenformin extended lifespan – this was a 1980 paper
- He did this again in a couple of additional short-lived mouse strains, also both cancer prone, in the early 2000s and saw that metformin extended lifespan
- At the same time, metformin was becoming quite a popular drug for diabetes treatment in people
- Epidemiological evidence from studies in people, where diabetics taking metformin appear certainly to live longer than diabetics not taking metformin but also maybe protected against a variety of other age-related diseases
Back to the mouse studies: Rafael de Cabo at NIA did a study of metformin in C57BL/6J six mice
- They actually tested two doses of metformin
- One dose, the low dose extended lifespan by about 5%.
- The higher dose shortened lifespan by 10%.
- Yet, the title of the paper was “Metformin Increases Lifespan and Health Span in Mice”
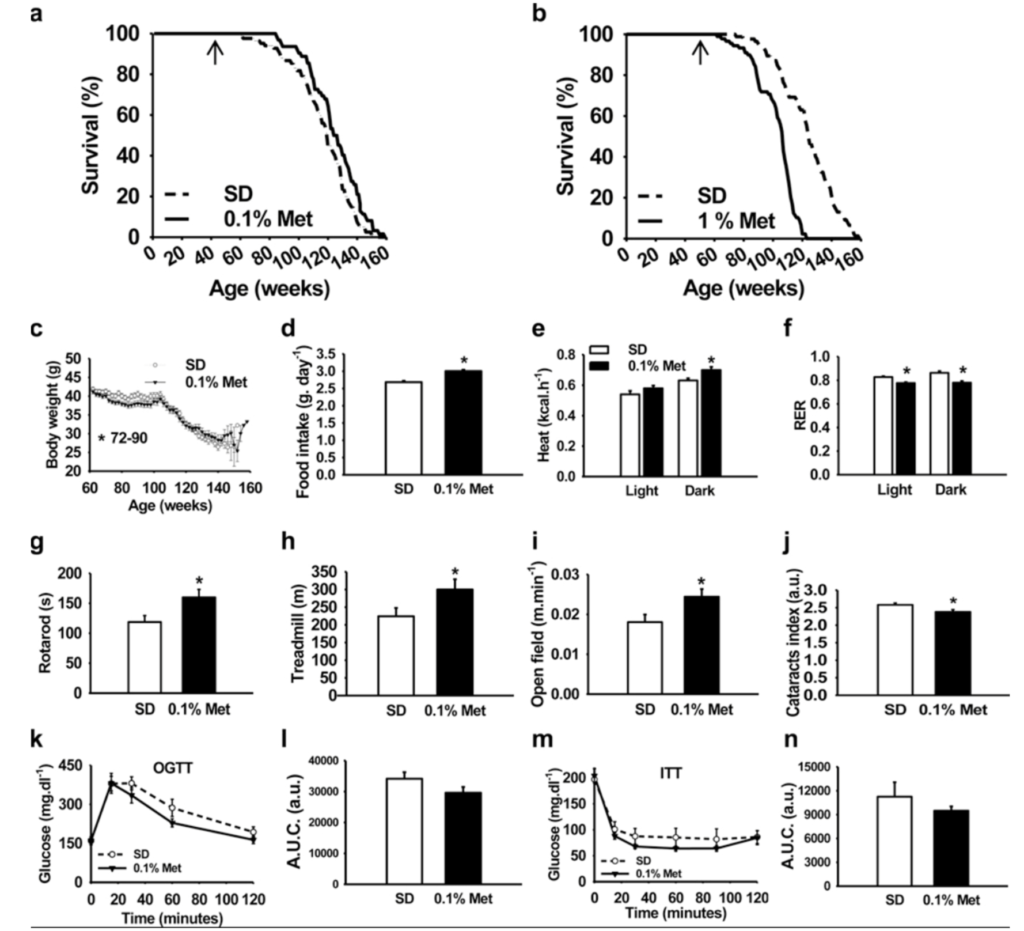
Figure 8. Source.
- On the upper left is the 0.1% metformin, small effect on lifespan, but statistically significant
- When we compared the control curves across four different studies, the controls in this study were pretty short-lived
- And then at the higher dose, a pretty substantial lifespan shortening
- So you could have just as easily said metformin shortens lifespan, but of course, the answer that they wanted to get was that metformin increases lifespan and that’s what ended up in the title.
- They do have some data in here that, at the lower dose, metformin can improve metabolic function, glucose tolerance, things like that
- So the claim was also made that metformin improves healthspan
- This is a pet peeve of Matt: “we should be very careful about claiming that an intervention increases healthspan when healthspan is not something that we can quantitatively measure”
- there is no set of quantitative measurements to assess health span and we need to be specific about the health span metrics that a given intervention can improve
Rapamycin vs. metformin
- The thing that maybe differentiates rapamycin from metformin somewhat is that, at least in mice, rapamycin really seems to broadly enhance function across many different tissues and organs
- That’s less clear with metformin, but certainly for glucose homeostasis and metabolic function, as we would expect from its use as an antidiabetic in people, metformin seems to be pretty protective against age-related changes in glucose homeostasis in mice
More about the metformin data
(Peter talked about this in the recent podcast with Nir Barzilai)
- The metformin observational study was comparing:
- i) diabetics with metformin against diabetics taking other medications and then
- ii) diabetics on metformin versus nondiabetics not on metformin
- In that study, you have cohorts that will demonstrate the superiority of the patients taking metformin even compared to the nondiabetics
- In other words, A diabetic with metformin on board seems to have more protection from cancer than a nondiabetic
- At first glance that sounds awfully convincing
- But when you really get into and scrutinize the methodology of those observational studies, you realize that there are some limitations and some biases
- First, the most obvious biases in the cohorts of diabetics is that, by definition, any patient whose diabetes progresses to the point where they need to add additional therapy, inclusive of insulin, really can’t be compared to a patient who is able to maintain glucose homeostasis on metformin alone
- Secondly, when you look at the survivorship bias in the patients taking metformin alone, you realize that a lot of patients get excluded from that type of analysis
- So if at the beginning of that cohort, you have all the patients who are on monotherapy metformin, and then what happens is:
- a subset of those will progress and require additional medication and they’re excluded from the analysis
- Another subset of those patients will not be able to remain compliant with their medications and they will be excluded from that analysis
- So you’re really concentrating the healthiest, most compliant patients
- “I’m not really convinced that that guarantees the insight we’d like to see there” says Peter
- All of this speaks to the importance of doing the TAME trial
How Peter uses metformin in patients:
- In Peter’s practice, he’s not recommending patients take NR or NMN, pending further data
- And he’s also not really using metformin as a geo-protective molecule
- He will prescribe metformin liberally in patients who are insulin resistant or even hyperinsulinemic in a subset of patients for whom he doesn’t believe he can get lots of aerobic exercise out of them
- That said, if he thinks that he can reverse their insulin resistance with exercise and nutrition, he’d actually rather use an SGLT2 inhibitor over metformin
- From what Peter is seeing in terms of mitochondrial function in the presence of metformin, it doesn’t make sense to him to go hardcore down the path of mitochondrial therapy, which is effectively zone two exercise while having metformin on board
Matt’s thoughts on metformin
- There are a couple things about metformin that give Matt pause as to how effective it’s going to be broadly for biology of aging
- One is the intervention testing program also did not detect a lifespan extension from metformin
- Secondly, data from Ben Miller’s lab suggesting that metformin may actually offset some of the benefits of exercise
- Thirdly , just like the NAD precursors, metformin’s a really “dirty drug”
Defining a “clean drug” and a “dirty drug” [1:38:00]
Dirty drugs
- This means that, biochemically, the mechanism of action is unclear
- For instance, we know there are multiple targets that metformin hits biochemically in cells, and NAD, as we’ve already alluded to, hits hundreds, if not thousands of different targets
- A dirty drug is something that’s nonspecific, that has several potential ways that it might be acting
- People still don’t know how metformin is working, from a mechanistic perspective, as a drug for diabetes
- There are several mechanisms that have been proposed, including
- activation of AMP kinase
- at least at higher doses, is an electron transport chain inhibitor
- It can affect folate metabolism by the microbiome
- In terms of effects on aging, it’s really unclear how effective metformin actually is in mice, but secondarily, what the mechanism of action would be.
- What Matt means by a “dirty drug” is it could be acting through any number of mechanisms and it’s hard to know whether you’ve got specificity in how it’s working
Clean drugs
A clean drug would be something, biochemically, that only hits one target.
Examples:
- Peter thinks in a “two by two” which is mechanism of action: known, unknown; Biomarker: present, absent
- One of the challenges of rapamycin is we don’t have a good biomarker, but at least we have a clear mechanism of action
- Conversely, metformin, we don’t really have a clear mechanism of action and we don’t have a good biomarker
- NR and NMN, we don’t have a good biomarker and we don’t have a clear mechanism of action
- The point here, on one level, is this gero-protective space is not that tidy
- It’s not like statins where you know exactly which enzyme is inhibited and have a perfect biomarker
- Matt responds, “I think it depends on the biomarker you’re talking about.”
- If you’re talking about a biomarker of efficacy for aging, he agrees that we don’t have that for anything because we don’t have any biomarkers of efficacy for aging
- But we do have clear biochemical biomarkers, certainly for rapamycin
- We know rapamycin is a very specific inhibitor of mTOR, and we have good biochemical ways to measure mTOR inhibition
- The challenge is we don’t know what level of mTOR inhibition is optimal for aging, and that gets back to the aging biomarkers
Reason for the lack of rapamycin studies in humans compared to NAD and metformin [1:41:00]
Rapamycin data
- As much as we have really convincing data of rapamycin’s life-extending capabilities in basically every species that it’s been tested in, we still don’t know exactly how to translate that to humans
- We indirectly have lots of ideas, but again, absent that biomarker, we don’t know how to make the link between mTORC1 inhibition and lifespan
Why there’s less studies for rapamycin in humans compared to NAD and metformin
Matt’s take on the lack of human trials in rapamycin:
- it’s a slow, frustrating process for Matt
- But we’ve got some ideas—People are coalescing around a general strategy for testing rapamycin in people
- If you go look on clinicaltrials.gov, you will probably find dozens of clinical trials of NAD precursors and many trials for metformin
- But you’ll find very few trials of rapamycin or sirolimus for the kinds of age-related indications where we actually think it could have an effect (even though the data, pre-clinically at least, are by far strongest for rapamycin)
- This is an unfortunate side effect of the way that clinical trials are done, the way that drugs are regulated
- A colleague of Matt’s got a grant to do a small clinical trial for periodontal disease for rapamycin because they have reason to believe rapamycin can reverse periodontal disease
- The bureaucratic hoops with FDA and in NID—even though this is an approved drug off label—are a huge barrier to people wanting to do these kinds of clinical trials
- Whereas it’s really easy to do a clinical trial for NAD precursors because they’re not regulated and pretty easy to do one for metformin because everybody believes it’s a safe drug
- People think of metformin as super safe even though, anecdotally, Matt’s heard more stories of side effects from people taking metformin off label than he has from people taking low-dose rapamycin once a week off label
Peter’s additional thoughts on what’s holding rapamycin back:
- One is the “unknown unknowns” versus the “known knowns”
- In Peter’s experience, metformin has tons of side effects
- If you push that dose hard enough, about 20% of people experience significant GI distress, for example
- Outside of aphthous ulcers, which tend to go away pretty quickly, Peter is not aware of a side effect with rapamycin in this pulsatile fashion
- “It does have a little bit of a mystery molecule shtick around it”
- The other issue, says Peter, is that rapamycin has almost no financial incentives for it to be studied in the way that there is a financial incentive for NR and NMN to be studied because there are all these proprietary blends, and there’s lots of commercial interest around it
Matt adds:
- For a long time, Matt has wondered why don’t academics do some of these clinical trials and he’s become convinced that a large part of it is the bureaucratic challenges with just being able to test rapamycin— “the hoops you have to jump through, as an academic, to get an IND approval or IND waiver, it’s ridiculous”
- Furthermore, when a drug or supplement gets studied over and over, it’s just a matter of time until you get some “positive” results which can mislead you for a long time
- On the flip side, a drug that’s only studied a handful of times, can come back “negative”—and there’s just many many reasons why that could happen—and that could halt the interests in it
Dasatinib and Quercetin as senolytics [1:47:00]
- Peter brings up something he’s been looking into recently, and that being a couple of really tiny studies that have suggested that Dasatinib and Quercetin—which Peter thinks is quite a toxic chemotherapeutic agent—but if given once a month, one day a month, can whack senescent cells
- There’s a tiny phase 1 study going on testing it in patients with mild cognitive impairment
- “But boy, it is pushing water up a hill to get these things done.” says Peter
Ranking the geroprotective molecules in terms of risk and reward [1:48:00]
Peter often uses an analogy that depicts the level of risk and reward when considering an action or behavior or new drug:
- Reward side of the equation:
- Are you picking up a penny?
- Are you picking up a gold coin?
- Risk side of the equation:
- Are you doing this in front of a slowly-rolling tricycle?
- Are you doing this in front of a speeding train?
- To look at the extremes…
- Nobody picks up pennies in front of trains
- But everybody would pick up a gold coin in front of a slowly-moving tricycle
- But this is really a continuum so the question becomes: What do you do in the middle ground?
In terms of reward, Peter would rank this molecules as follows:
- Based on current knowledge, rapamycin has more reward than the others
- Second would be metformin with the caveat that it’s likely to have a much bigger reward in an insulin-resistant person than an insulin-sensitive person
- Next would be NR and NMN (and he cannot distinguish between NMN and NR, at least based on the published data)
In terms of risk, Peter has the following thoughts:
- There’s the potentially esoteric risk of NR or NMN in a patient who has cancer, be it subclinical or otherwise, very theoretical. But note that there’s nothing in the human data to substantiate this
- Metformin has lots of side effects—it can be a miserable drug to take, but Peter doesn’t think it’s a particularly risky drug
- With rapamycin, at the surface, some would be concerned about immune suppression
- But at the doses we’re talking about, in a pulsatile fashion, it actually seems to enhance the immune system
Overall thoughts on risk:
- Peter actually sees of these molecules being much closer to the “tricycle side of the equation”
- You have to ask, risky in contrast to what?
“Life is a dangerous thing. We’re all inevitably marching towards death. One has to at least be somewhat prepared to take some risk in order to mitigate the risk of death.” —Peter Attia
What about healthspan and quality of life?
- When you’re talking about risk and reward, it’s not only about death, it’s about quality of life
- metformin can be a miserable drug to take if you have gastrointestinal distress or other side effects that affects your quality of life
- Same thing is true on the potential reward side
- Matt would put rapamycin at the top of that list in terms of reward
- Personally, Matt had very severe pain in his shoulder, he had adhesive capsulitis, and he believes that rapamycin cured that for him which improved his quality of life
- Those other aspects that influence how you feel and how much energy you have are really important to consider as well
- Matt admits he’s biased towards rapamycin and it’s because of how closely he works with the data and the people he’s speaks to about their experience
“I’ve seen the data, I’ve done a lot of the experiments, and I’ve talked to a lot of people, and I’m pretty convinced that for a lot of people, rapamycin can have a positive effect on their quality of life.” —Matt Kaeberlein
Selected Links / Related Material
AMA from February 2021 where Bob and Peter tried to simplify the complex topic of insulin: #149 – AMA #20: Simplifying the complexities of insulin resistance: how it’s measured, how it manifests in the muscle and liver, and what we can do about it | Peter Attia (peterattiamd.com) [3:45]
- The questions answer in the AMA came out of the conversation with Gerry Shulman: #140 – Gerald Shulman, M.D., Ph.D.: A masterclass on insulin resistance—molecular mechanisms and clinical implications | Peter Attia (peterattiamd.com)
Previous podcasts on topics related to this AMA #35 episode: [4:30]
- Episode with Matt Kaeberlein:
- Episode with Steve Austad: #171 – Steve Austad, Ph.D.: The landscape of longevity science: making sense of caloric restriction, biomarkers of aging, and possible geroprotective molecules [4:30]
- Episodes with Nir Barzilai:
- Episode with Joan Mannick: #123 – Joan Mannick, M.D. & Nir Barzilai, M.D.: Rapamycin and metformin—longevity, immune enhancement, and COVID-19
- Episodes with David Sinclair:
- Episode with Lloyd Klickstein: #118 – Lloyd Klickstein, M.D., Ph.D.: Rapamycin, mTOR inhibition, and the biology of aging | Peter Attia (peterattiamd.com) [4:30]
- Episode with David Sabatini: #09 – David Sabatini, M.D., Ph.D.: rapamycin and the discovery of mTOR — the nexus of aging and longevity? [4:30]
Matt’s project testing rapamycin in pet dogs: Dog Aging Project | (dogagingproject.org) [31:15]
One example of data that reports that rhesus monkeys can live longer with calorie restriction: Caloric restriction improves health and survival of rhesus monkeys (Mattison et al., 2017) [33:00]
Homepage for the Interventions Testing Program: Interventions Testing Program (ITP) | (nia.nih.gov) [36:30]
Matt’s 2016 paper testing rapamycin in mice: Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice (Bitto et al., 2016) [44:15, 1:33:00]
Rich Miller episode of The Drive discussing the Interventions Testing Program: #148 – Richard Miller, M.D., Ph.D.: The gold standard for testing longevity drugs: the Interventions Testing Program | Peter Attia (peterattiamd.com) [45:00]
ITP studies of rapamycin in mice show lifespan extension: [45:00]
2000 paper looking at NAD and the ability of caloric restriction to extend lifespan in yeast: Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae (Lin et al., 2000) [55:00]
Matt’s paper that suggested that lifespan extension from caloric restriction was independent of SIR-2: Sir2-independent life span extension by calorie restriction in yeast (Kaeberlein et al., 2004) [1:01:15]
ITP study of NR that failed to extend lifespan in mice: The Latest Data from the Interventions Testing Program: Nicotinamide Riboside has No Effect on Mouse Life Span | (fightaging.org) [1:11:30]
Study that reported that late life administration of nicotinamide riboside in C57 black 6 mice had a small, but statistically significant effect on survival: NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice (Zhang et al., 2016) [1:11:30]
Study by Vilhelm Bohr showing where NAD precursors could be effective such as in situations where there is high levels of DNA damage: NAD+ replenishment improves lifespan and healthspan in Ataxia telangiectasia models via mitophagy and DNA repair (Fang et al., 2016) [1:19:00]
Episode of The Drive with Inigo San Milan where he briefly discussed the data about NR being potentially associated with increased cancer risk: #201 – Deep dive back into Zone 2 | Iñigo San-Millán, Ph.D. (Pt. 2) | Peter Attia (peterattiamd.com) [1:23:15]
The ALS study—one of the few human studies using NAD precursors: Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled human pilot study (de la Rubia, et al., 2019) [1:26:00]
Very small human study with NAD precursors published in Science: Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women (Yoshino et al., 2021) [1:27:00]
The first studies of metformin for lifespan actually sprang out of earlier studies of phenformin done by a Russian scientist named Oleg Anisimov: Effect of treatment with phenformin, diphenylhydantoin or L-dopa on life span and tumour incidence in C3H/Sn mice (Dilman and Anisimov, 1980) [1:29:15]
Metformin study by Anisimov showing that metformin extended lifespan in cancer prone, short-lived mouse strains: Metformin slows down aging and extends life span of female SHR mice (Anisimov et al., 2008) [1:30:00]
Metformin paper with controversial title that suggested metformin increased lifespan in mice despite the data not fully backing that conclusion: Metformin improves healthspan and lifespan in mice (Martin-Montalvo et al., 2013) [1:31:15]
Episode of The Drive with Nir Barzilai discussing the latest research into metformin: #204 – Centenarians, metformin, and longevity | Nir Barzilai, M.D. | Peter Attia (peterattiamd.com) [1:33:45]
ITP study of metformin which did not show lifespan extension: Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α‐glucosidase inhibitor or a Nrf2‐inducer (Strong et al., 2016) [1:37:15]
Data from Ben Miller’s lab suggesting that metformin may actually offset some of the benefits of exercise: Metformin inhibits mitochondrial adaptations to aerobic exercise training in older adults (Konopka et al., 2019) [1:37:30]
People Mentioned
- Gerald Shulman [4:00]
- Steve Austad [4:30]
- Nir Barzilai [4:30, 1:33:45]
- Joan Mannick [4:30]
- David Sinclair [4:30]
- Lloyd Klickstein [4:30]
- David Sabatini [4:30]
- Rich Miller [45:00]
- Leonard Guarente [55:00]
- Su-Ju Lin [55:30]
- Josh Rabinowitz [1:14:00]
- Vilhelm Bohr 1:19:00]
- Inigo San Millan [1:23:15]
- Oleg Anisimov [1:29:15]
- Rafael de Cabo [1:31:15]
- Ben Miller [1:37:30]