
Want to catch up with other articles from this series?
- The straight dope on cholesterol – Part I
- The straight dope on cholesterol – Part II
- The straight dope on cholesterol – Part III
- The straight dope on cholesterol – Part IV
- The straight dope on cholesterol – Part V
- The straight dope on cholesterol – Part VI
- The straight dope on cholesterol – Part VII
- The straight dope on cholesterol – Part VIII
- The straight dope on cholesterol – Part IX
Previously, in Part I, Part II, Part III, Part IV, Part V ,and Part VI of this series, we addressed these 8 concepts:
#1 — What is cholesterol?
#2 — What is the relationship between the cholesterol we eat and the cholesterol in our body?
#3 — Is cholesterol bad?
#4 — How does cholesterol move around our body?
#5 – How do we measure cholesterol?
#6 – How does cholesterol actually cause problems?
#7 – Does the size of an LDL particle matter?
#8 – Why is it necessary to measure LDL-P, instead of just LDL-C?
(No so) Quick refresher on take-away points from previous posts, should you need it:
- Cholesterol is “just” another fancy organic molecule in our body but with an interesting distinction: we eat it, we make it, we store it, and we excrete it – all in different amounts.
- The pool of cholesterol in our body is essential for life. No cholesterol = no life.
- Cholesterol exists in 2 forms – unesterified or “free” (UC) and esterified (CE) – and the form determines if we can absorb it or not, or store it or not (among other things).
- Much of the cholesterol we eat is in the form of CE. It is not absorbed and is excreted by our gut (i.e., leaves our body in stool). The reason this occurs is that CE not only has to be de-esterified, but it competes for absorption with the vastly larger amounts of UC supplied by the biliary route.
- Re-absorption of the cholesterol we synthesize in our body (i.e., endogenous produced cholesterol) is the dominant source of the cholesterol in our body. That is, most of the cholesterol in our body was made by our body.
- The process of regulating cholesterol is very complex and multifaceted with multiple layers of control. I’ve only touched on the absorption side, but the synthesis side is also complex and highly regulated. You will discover that synthesis and absorption are very interrelated.
- Eating cholesterol has very little impact on the cholesterol levels in your body. This is a fact, not my opinion. Anyone who tells you different is, at best, ignorant of this topic. At worst, they are a deliberate charlatan. Years ago the Canadian Guidelines removed the limitation of dietary cholesterol. The rest of the world, especially the United States, needs to catch up. To see an important reference on this topic, please look here.
- Cholesterol and triglycerides are not soluble in plasma (i.e., they can’t dissolve in water) and are therefore said to be hydrophobic.
- To be carried anywhere in our body, say from your liver to your coronary artery, they need to be carried by a special protein-wrapped transport vessel called a lipoprotein.
- As these “ships” called lipoproteins leave the liver they undergo a process of maturation where they shed much of their triglyceride “cargo” in the form of free fatty acid, and doing so makes them smaller and richer in cholesterol.
- Special proteins, apoproteins, play an important role in moving lipoproteins around the body and facilitating their interactions with other cells. The most important of these are the apoB class, residing on VLDL, IDL, and LDL particles, and the apoA-I class, residing for the most part on the HDL particles.
- Cholesterol transport in plasma occurs in both directions, from the liver and small intestine towards the periphery and back to the liver and small intestine (the “gut”).
- The major function of the apoB-containing particles is to traffic energy (triglycerides) to muscles and phospholipids to all cells. Their cholesterol is trafficked back to the liver. The apoA-I containing particles traffic cholesterol to steroidogenic tissues, adipocytes (a storage organ for cholesterol ester) and ultimately back to the liver, gut, or steroidogenic tissue.
- All lipoproteins are part of the human lipid transportation system and work harmoniously together to efficiently traffic lipids. As you are probably starting to appreciate, the trafficking pattern is highly complex and the lipoproteins constantly exchange their core and surface lipids.
- The measurement of cholesterol has undergone a dramatic evolution over the past 70 years with technology at the heart of the advance.
- Currently, most people in the United States (and the world for that matter) undergo a “standard” lipid panel, which only directly measures TC, TG, and HDL-C. LDL-C is measured or most often estimated.
- More advanced cholesterol measuring tests do exist to directly measure LDL-C (though none are standardized), along with the cholesterol content of other lipoproteins (e.g., VLDL, IDL) or lipoprotein subparticles.
- The most frequently used and guideline-recommended test that can count the number of LDL particles is either apolipoprotein B or LDL-P NMR, which is part of the NMR LipoProfile. NMR can also measure the size of LDL and other lipoprotein particles, which is valuable for predicting insulin resistance in drug naïve patients, before changes are noted in glucose or insulin levels.
- The progression from a completely normal artery to a “clogged” or atherosclerotic one follows a very clear path: an apoB containing particle gets past the endothelial layer into the subendothelial space, the particle and its cholesterol content is retained, immune cells arrive, an inflammatory response ensues “fixing” the apoB containing particles in place AND making more space for more of them.
- While inflammation plays a key role in this process, it’s the penetration of the endothelium and retention within the endothelium that drive the process.
- The most common apoB containing lipoprotein in this process is certainly the LDL particle. However, Lp(a) and apoB containing lipoproteins play a role also, especially in the insulin resistant person.
- If you want to stop atherosclerosis, you must lower the LDL particle number. Period.
- At first glance it would seem that patients with smaller LDL particles are at greater risk for atherosclerosis than patients with large LDL particles, all things equal.
- “A particle is a particle is a particle.” If you don’t know the number, you don’t know the risk.
- With respect to laboratory medicine, two markers that have a high correlation with a given outcome are concordant – they equally predict the same outcome. However, when the two tests do not correlate with each other they are said to be discordant.
- LDL-P (or apoB) is the best predictor of adverse cardiac events, which has been documented repeatedly in every major cardiovascular risk study.
- LDL-C is only a good predictor of adverse cardiac events when it is concordant with LDL-P; otherwise it is a poor predictor of risk.
- There is no way of determining which individual patient may have discordant LDL-C and LDL-P without measuring both markers.
- Discordance between LDL-C and LDL-P is even greater in populations with metabolic syndrome, including patients with diabetes. Given the ubiquity of these conditions in the U.S. population, and the special risk such patients carry for cardiovascular disease, it is difficult to justify use of LDL-C, HDL-C, and TG alone for risk stratification in all but the most select patients.
- To address this question, however, one must look at changes in cardiovascular events or direct markers of atherosclerosis (e.g., IMT) while holding LDL-P constant and then again holding LDL size constant. Only when you do this can you see that the relationship between size and event vanishes. The only thing that matters is the number of LDL particles – large, small, or mixed.
Concept #9 – Does “HDL” matter after all?
Last week was the largest annual meeting of the National Lipid Association (NLA) in Phoenix, AZ. The timing of the meeting could not have been better, given the huge buzz going around on the topic of “HDL.” (If you’re wondering why I’m putting HDL in quotes, I’ll address it shortly.)
What buzz, you ask? Many folks, including our beloved health columnists at The New York Times, are talking about the death of the HDL hypothesis – namely, the notion that HDL is the “good cholesterol.”
Technically, this “buzz” started about 6 years ago when Pfizer made headlines with a drug in their pipeline called torcetrapib. Torcetrapib was one of the most eagerly anticipated drugs ever, certainly in my lifetime, as it had been shown to significantly raise plasma levels of HDL-C. You’ll recall from part II of this series, HDL particles play an important role in carrying cholesterol from the subendothelial space back to the liver via a process called reverse cholesterol transport (RCT). Furthermore, many studies and epidemiologic analyses have shown that people with high plasma levels of HDL-C have a lower incidence of coronary artery disease.
In the case of torcetrapib, there was an even more compelling reason to be optimistic. Torcetrapib blocked the protein cholesterylester transfer protein, or CETP, which facilitates the collection and one-to-one exchange of triglycerides and cholesterol esters between lipoproteins. Most (but not all) people with a mutation or dysfunction of this protein were known to have high levels of HDL-C and lower risk of heart disease. Optimism was very high that a drug like torcetrapib, which could mimic this effect and create a state of more HDL-C and less LDL-C, would be the biggest blockbuster drug ever.
The past month or so has seen this discussion intensify, which I’ll quickly try to cover below.
The data
Torcetrapib
After several smaller clinical trials showed that patients taking torcetrapib experienced both an increase in HDL-C and a reduction in LDL-C, a large clinical trial pitting atorvastatin (Lipitor) against atorvastatin + torcetrapib was underway. This trial was to be the jewel in the crown of Pfizer. It was already known that Lipitor reduced coronary artery disease (and reduced LDL-C, though this may have been a bystander effect and real reduction in mortality may be better attributed to the reduction in LDL-P).
I still remember exactly where I was standing, on the corner of Kerney St. and California St. in the heart of San Francisco’s financial district, on that December day back in 2006 when it was announced the trial had been halted because of increased mortality in the group receiving torcetrapib. In other words, adding torcetrapib actually made things worse. I was shocked.
Many reasons were offered for this, including the notion that torcetrapib was, indeed, helpful, but because of unanticipated side-effects, (raising blood pressure in some patients and altering electrolyte balance in others), the net impact was harmful. Some even suggested that the drug could be useful in the “right” patients (e.g., those with low HDL-C, but normal blood pressure). Furthermore, in two subsequent studies looking at carotid IMT (thickening of the carotid arteries) and intravascular ultrasound, there was no reduction in atherosclerosis.
This was a big strike against the HDL hypothesis and work on torcetrapib was immediately halted.
Niacin
Niacin has long been known to raise HDL-C and has actually been used therapeutically for this reason for many years. The AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides – you can’t have trials in medicine without catchy names!) sought to test this. The trial randomly assigned over 3,000 patients with known and persistent, but stable and well treated cardiovascular risk, to one of two treatments:
- Simvastatin (40-80 mg/day), +/- ezetimibe (10 mg/day) as necessary to maintain LDL-C below 70 mg/dL + placebo (a tiny dose of crystalline niacin to cause flushing);
- As above, but instead of a placebo, patients were given 1,500 to 2,000 mg/day of extended-release niacin.
Both arms of the study had their LDL-C < 70 mg/dL, non-HDL-C < 100 md/dL and apoB < 80 mg/dL, but despite the statin or statin + ezetimibe treatment still had low HDL-C. So, if niacin raised HDL-C and reduced events, the HDL raising hypothesis would be proven.
Simvastatin, as its name suggests, is a statin which primarily works by blocking HMG-CoA reductuse, an enzyme necessary to synthesize endogenous cholesterol. Ezetimibe works on the other end of problem, by blocking the NPC1L1 transporter on gut enterocytes and hepatocytes at the hepatobiliary junction (for a quick refresher, go back to part I of this series and look at the second figure – ezetimibe blocks the “ticket taker” in the bar).
After two years the niacin group, as expected, had experienced a significant increase in plasma HDL-C (along with some other benefits like a greater reduction in plasma triglycerides). However, there was no improvement in patient survival. The trial was futile and the data and safety board halted the trial. In other words, for patients with cardiac risk and LDL-C levels at goal with medication niacin, despite raising HDL-C and lowering TG, did nothing to improve survival. This was another strike against the HDL hypothesis.
Dalcetrapib
By 2008, as the AIM-HIGH trial was well under way, another pharma giant, Roche, was well into clinical trials with another drug that blocked CETP. This drug, a cousin of torcetrapib called dalcetrapib, albeit a weaker CETP-inhibitor, appeared to do all the “right” stuff (i.e., it increased HDL-C) without the “wrong” stuff (i.e., it did not appear to adversely affect blood pressure). It did nothing to LDL-C or apoB.
This study, called dal-OUTCOMES, was similar to the other trials in that patients were randomized to either standard of care plus placebo or standard of care plus escalating doses of dalcetrapib. A report of smaller safety studies (called dal-Vessel and Dal-Plaque) was published a few months ago in the American Heart Journal, and shortly after Roche halted the phase 3 clinical trial. Once again, patients on the treatment arm did experience a significant increase in HDL-C, but failed to appreciate any clinical benefit. Another futile trial.
Currently, two additional CETP inhibitors, evacetrapib (manufactured by Lilly) and anacetrapib (manufactured by Merck) are being evaluated. They are much more potent CETP inhibitors and, unlike dalcetrapib, also reduce apoB and LDL-C and Lp(a). Both Lilly and Merck are very optimistic that their variants will be successful where Pfizer’s and Roche’s were not, for a number of reasons including greater anti-CETP potency.
Nevertheless, this was yet another strike against the HDL hypothesis because the drug only raised HDL-C and did nothing to apoB. If simply raising HDL-C without attacking apoB is a viable therapeutic strategy, the trial should have worked. We have been told for years (by erroneous extrapolation from epidemiologic data) that a 1% rise in HDL-C would translate into a 3% reduction in coronary artery disease. These trials would suggest otherwise.
Mendelian randomization
On May 17 of this year a large group in Europe (hence the spelling of randomization) published a paper in The Lancet, titled, “Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study.” Mendelian randomization, as its name sort of suggests, is a method of using known genetic differences in large populations to try to “sort out” large pools of epidemiologic data.
In the case of this study, pooled data from tens of studies where patients were known to have myocardial infarction (heart attacks) were mapped against known genetic alterations called SNPs (single nucleotide polymorphisms, pronounced “snips”). I’m not going to go into detail about the methodology because it would take 3 more blog posts., But, the reason for doing this analysis was to ferret out if having a high HDL-C was (only) correlated with better cardiovascular outcome, which has been the classic teaching, or if there was any causal relationship. In other words, does having a high HDL-C cause you to have a lower risk of heart disease or is it a marker for something else?
This study found, consistent with the trials I’ve discussed above, that any genetic polymorphism that seems to raise HDL-C does not seem to protect from heart disease. That is, patients with higher HDL-C due to a known genetic alteration did not seem to have protection from heart disease as a result of that gene. This suggests that people with high or low HDL-C who get coronary artery disease may well have something else at play.
Oh boy. This seems like the last nail in the casket of the entire “HDL” hypothesis, as evidenced by all of the front page stories worldwide.
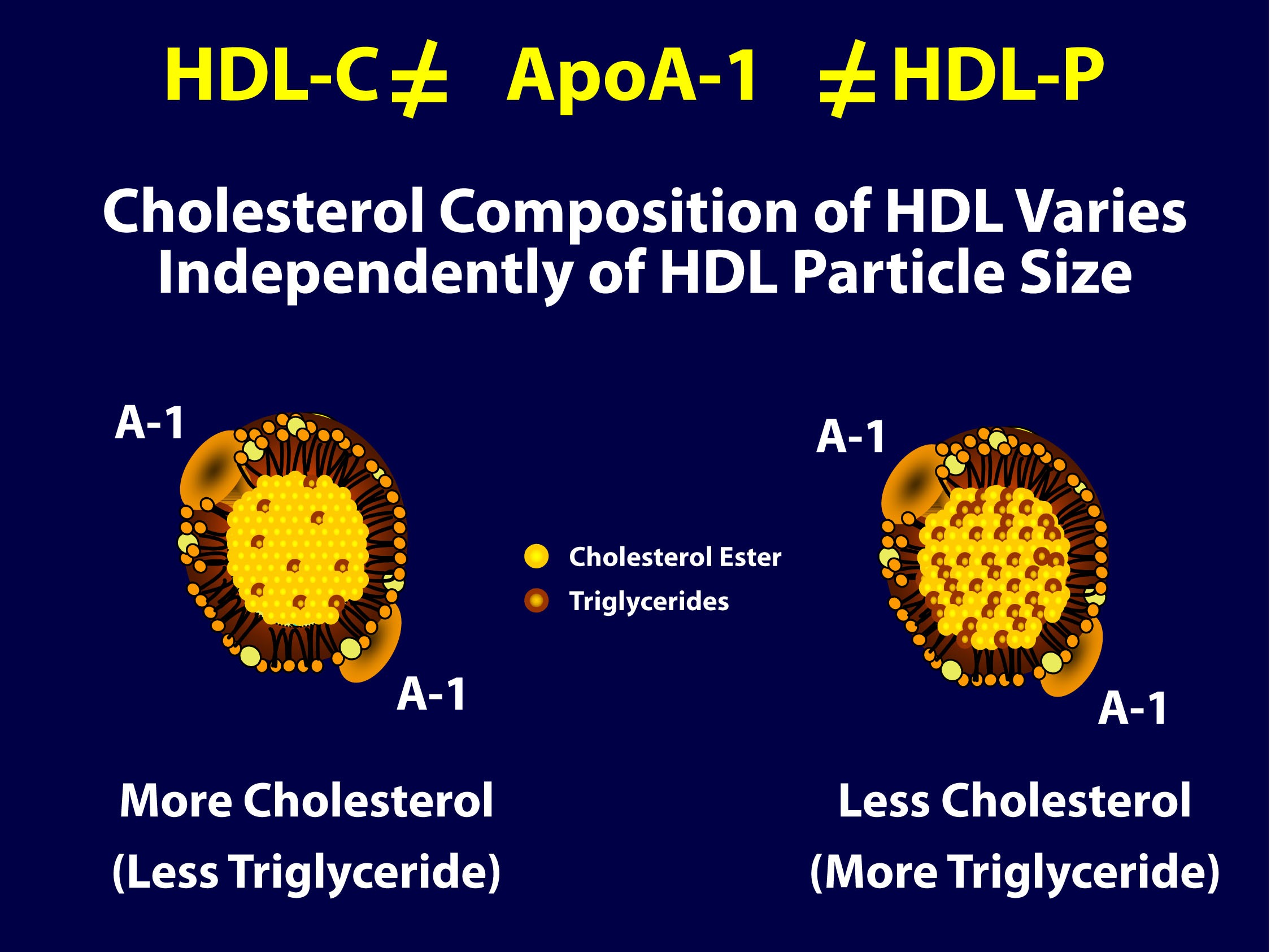
The rub: the difference between HDL-C and HDL-P
The reason I’ve been referring to high density lipoprotein as “HDL,” unless specifically referring to HDL-C, is that HDL-P and HDL-C are not the same thing. Just as you are now intimately familiar with the notion that LDL-C and LDL-P are not the same thing, the same is true for “HDL” which simply stands for high density lipoprotein, and like LDL is not a lab assay. In fact, unpublished data from the MESA trial found that the correlation between HDL-C and HDL-P was only 0.73, which is far from “good enough” to say HDL-C is a perfect proxy for HDL-P.
HDL-C, measured in mg/dL (or mmol/L outside of the U.S.), is the mass of cholesterol carried by HDL particles in a specified volume (typically measured as X mg of cholesterol per dL of plasma). HDL-P is something entirely different. It’s the number of HDL particles (minus unlipidated apoA-I and prebeta-HDLs: at most 5% of HDL particles) contained in a specified volume (typically measured as Y micromole of particles per liter).
As you can see in the figure below (courtesy of Jim Otvos’ presentation at the NLA meeting 2 weeks ago), the larger an HDL particle, the more cholesterol it carries. So, an equal number of large versus small HDL particles (equal HDL-P) can carry very different amounts of cholesterol (different HDL-C). Of course, it’s never this simple because HDL particles, like their LDL counterparts, don’t just carry cholesterol. They carry triglycerides, too. Keep in mind, HDL core CE/TG ratio is about 10:1 or greater – if the large HDL carries TG, it will not be carrying very much cholesterol.

So, the important point is that HDL-C is not the same as HDL-P (which is also not the same as apoAI, as HDL particles can carry more than one apoAI).
But there’s something else going on here. If you look at the figure below, from the Framingham cohort, you’ll note something interesting. As HDL-C rises, it does so not in a uniform or “across the board” fashion. A rise in HDL-C seems to disproportionately result from an increase in large HDL particles. In other words, as HDL-C rises, it doesn’t necessarily mean HDL-P is rising at all, and certainly not as much.
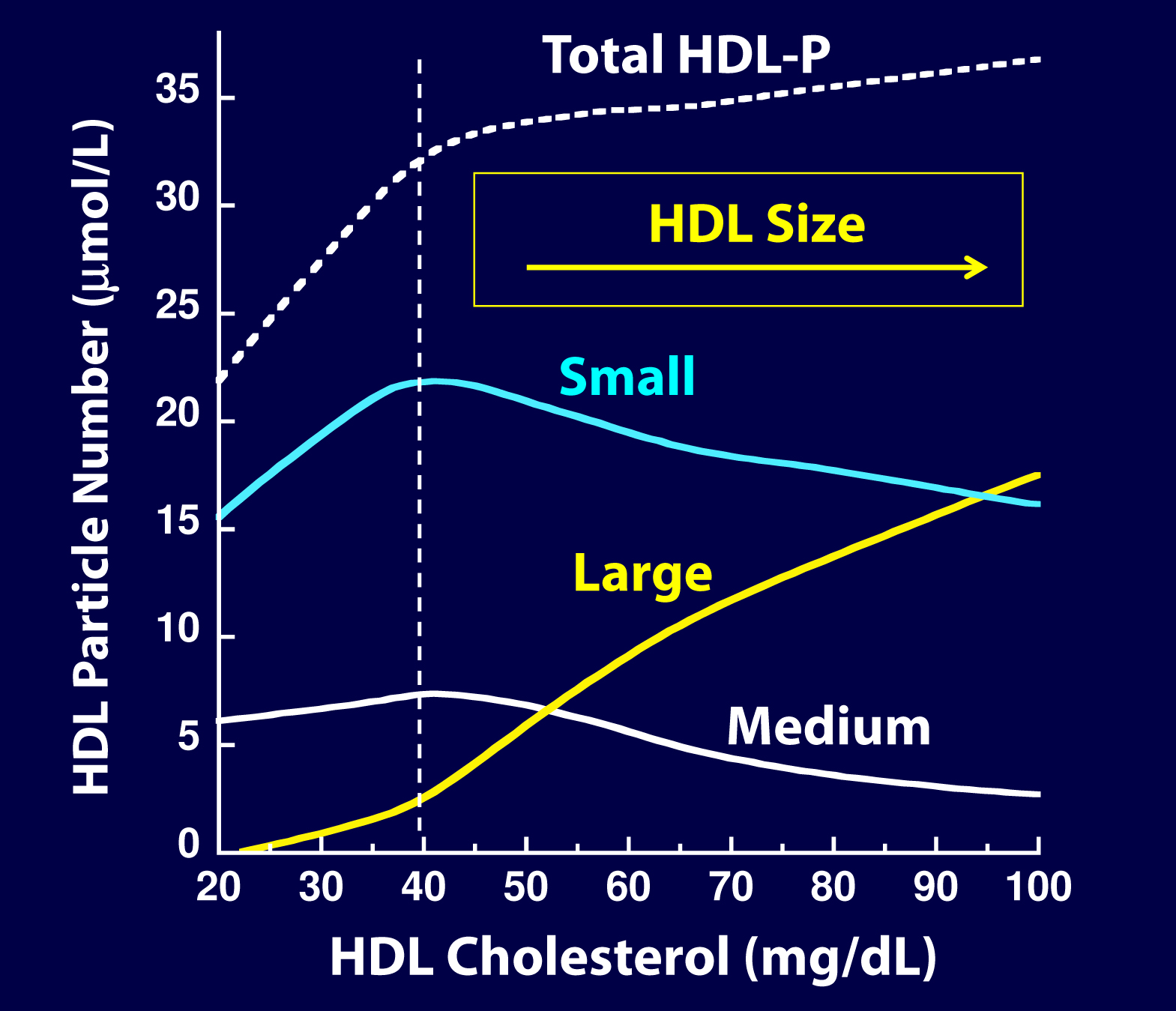
As you can see, for increases in HDL-C at low levels (i.e., below 40 mg/dL) the increase in small particles seems to account for much of the increase in total HDL-P, While for increases over 40 mg/dL, the increase in large particles seems to account for the increase in HDL-C. Also note that as HDL-C rises above 45 mg/dL, there is almost no further increase in total HDL-P – the rise in HDL-C is driven by enlargement of the HDL particle – more cholesterol per particle – not the drop in small HDL-P. This reveals to us that the small HDL particles are being lipidated.
Is there a reason to favor small HDL particles over large ones?
In the 2011 article, “Biological activities of HDL subpopulations and their relevance to cardiovascular disease,” published in Trends in Molecular Medicine, the authors describe in great detail some of protective mechanisms imparted by HDL particles.
Large HDL particles may be less protective and even dysfunctional in certain pathological states, whereas small to medium-sized HDL particles seem to confer greater protection through the following mechanisms:
- Greater antioxidant activity
- Greater anti-inflammatory activity
- Greater cholesterol efflux capacity
- Greater anti-thrombotic properties
In other words, particle for particle, it seems a small HDL particle may be better at transporting cholesterol from the subendothelial space (technically, they acquire cholesterol from cholesterol-laden macrophages or foam cells in the subedothelial space) elsewhere, better at reducing inflammation, better at preventing clotting, and better at mitigating the problems caused by oxidative free radicals.
Of course, reality is complicated. If there was no maturation from small to large HDL particles (i.e., the dynamic remodeling of HDL), the system would be faulty. So, the truth is that all HDL sizes are required and that HDL particles are in a constant dynamic state (or “flux”) of lipidating and delipidating, and the real truth is no particular HDL size can be said to be the best. If the little HDLs do not enlarge, the ApoA-I mediated lipid trafficking system is broken.
The truth about the old (and overly simplistic) term called reverse cholesterol transport (RCT)
HDL particles traffic cholesterol and proteins and last in plasma on average for 5 days. They are in a constant state of acquiring cholesterol (lipidation) and delivering cholesterol (delipidation). There are membrane receptors on cells that can export cholesterol to HDL particles (sterol efflux transporters) or extract cholesterol or cholesterol ester from HDL particles (sterol influx transporters).
The vast majority of lipidation occurs (in order): 1) at the liver, 2) the small intestine, 3) adipocytes and 4) peripheral cells, including plaque if present. The liver and intestine account for 95% of this process. The amount of cholesterol pulled out of arteries (called macrophage reverse cholesterol transport) is critical to disease prevention but is so small it has no effect on serum HDL levels. Even in patients with extensive plaque, the cholesterol in that plaque is about 0.5% of total body cholesterol. HDL particles circulate for several days as a ready reserve of cholesterol: almost no cell in humans require a delivery of cholesterol as cells synthesize all they need. However, steroidogenic hormone producing tissues (e.g., adrenal cortex and gonads) do require cholesterol and the HDL particle is the primary delivery truck.
If, as is the case in a medical emergency, the adrenal gland must rapidly make a lot of cortisone, the HDL particles are there with the needed cholesterol. This explains the low HDL-C typically seen in patients with severe infections (e.g., sepsis) and severe inflammatory conditions (e.g., Rheumatoid Arthritis).
Sooner or later HDL particles must be delipidated, and this takes place at: 1) the adrenal cortex or gonads 2) the liver, 3) adipocytes, 4) the small intestine (TICE or transintestinal cholesterol efflux) or give its cholesterol to an apoB particle (90% of which are LDLs) to return to the liver. A HDL particle delivering cholesterol to the liver or intestine is called direct reverse cholesterol transport (RCT), whereas a HDL particle transferring its cholesterol to an apoB particle which returns it to the liver is indirect RCT. Hence, total RCT = direct RCT + indirect RCT.
The punch line: a serum HDL-C level has no known relationship to this complex process of RCT. The last thing a HDL does is lose its cholesterol. The old concept that a drug or lifestyle that raises HDL-C is improving the RCT process is wrong; it may or may not be affecting that dynamic process. Instead of calling this RCT, it would be more appropriately called apoA-I trafficking of cholesterol.
Why do drugs that specifically raise HDL-C seem to be of little value?
As I’ve argued before, while statins are efficacious at preventing heart disease, it’s sort of by “luck” as far as most prescribing physicians are concerned. Most doctors use cholesterol lowing medication to lower LDL-C, not LDL-P. Since there is an overlap (i.e., since the levels of LDL-P and LDL-C are concordant) in many patients, this misplaced use of statins seems to work “ok.” I, and many others far more knowledgeable, would argue that if statins and other drugs were used to lower LDL-P (and apoB), instead of LDL-C, their efficacy would be even greater. The same is true for dietary intervention.
Interestingly, (and I would have never known this had Jim Otvos not graciously spent a hour on the phone with me two weeks ago giving me a nuanced HDL tutorial), a study that went completely unnoticed by the press in 2010, published in Circulation, actually did a similar analysis to the Lancet paper, except that the authors looked at HDL-P instead of HDL-C as the biomarker and looked at the impact of phospholipid transfer protein (PLTP) on HDL metabolism. In this study, though not the explicit goal, the authors found that an increase in the number of HDL particles and smaller HDL particles decreased the risk of cardiovascular disease. The key point, of course, is that the total number of HDL particles rose, and it was driven by increased small HDL-P. The exact same thing was seen in the VA-HIT trial: the cardiovascular benefit of the treatment (fibrate) was related to the rise in total HDL-P which was driven by the fibrates’ ability to raise small HDL-P.
It seems the problem with the “HDL hypothesis” is that it’s using the wrong marker of HDL. By looking at HDL-C instead of HDL-P, these investigators may have missed the point. Just like LDL, it’s all about the particles.
Summary
- HDL-C and HDL-P are not measuring the same thing, just as LDL-C and LDL-P are not.
- Secondary to the total HDL-P, all things equal it seems smaller HDL particles are more protective than large ones.
- As HDL-C levels rise, most often it is driven by a disproportionate rise in HDL size, not HDL-P.
- In the trials which were designed to prove that a drug that raised HDL-C would provide a reduction in cardiovascular events, no benefit occurred: estrogen studies (HERS, WHI), fibrate studies (FIELD, ACCORD), niacin studies, and CETP inhibition studies (dalcetrapib and torcetrapib). But, this says nothing of what happens when you raise HDL-P.
- Don’t believe the hype: HDL is important, and more HDL particles are better than few. But, raising HDL-C with a drug isn’t going to fix the problem. Making this even more complex is that HDL functionality is likely as important, or even more important, than HDL-P, but no such tests exist to “measure” this.
Two apolipoprotein A1 chains (magenta ribbons) complexed with cholesterol (orange balls) and phospholipids, after PDB 3K2S by Ayacop [Public domain], via Wikimedia Commons

{kind=link}
Thank you again for another cholesterol post Dr. Attia! I’ve been referring my friends and peers to your site because of it.
I also made a short cholesterol comic based on some of your ideas and Dr. Dayspring’s lectures to try and pull people into these newer ideas who may be intimidated by the details. If interested, you can see it here: https://thefatnurse.wordpress.com/2012/06/06/cholesterol-for-all-ages/
Once again, thank you for this series. You and your blog are doing a great service!
Yes, I did see your fantastic cartoons. I’m honored to be a small part of your journey into the world of lipidology. I hope folks take a moment to check out your link. I really enjoyed it.
Fun comic! I like the idea of a low carb diet encouraging cholesterol to carpool (for some people).
Sounds like the optimal situation would be most LDL cholesterol bits packed into big buses, and most HDL cholesterol bits riding in low occupancy sports cars.
This blog just keeps on giving!
The comics are GREAT! Thanks FatNurse and thanks Peter!
Really great comic. Hope it’s ok that I shared it on Facebook.
And Peter, who does your art (like that nifty graphic of the HDL mop)?
Great “Comic”! There’s a typo on page 12 in the 4th frame: “lead” should be “led.”
Thanks for the kind words! I’ve always wondered how Dr. Attia has so much time and energy to read and write all his insightful posts while starting his other side projects….but now I know: its powered by pure passion.
This whole area of study is so fascinating that I find myself reading literature and reaching out to people in the field in my spare time for…fun lol. I only hope that my cartoons can pique the interest of others who have been too influenced by the low fat dogma to bother looking at anything else.
And yes lorraine feel free to share my comic on face book. I’ll be adding more “…for all ages” comics in the future as well.
Thank you for the post Peter. I doubt the lay press will have the wherewithal to understand/communicate any of this effectively, so I fear they will throw the HDL-P out with the HDL-C bathwater. Then again now that I think about it, I’m not all that sanguine about the ability of the scientific journals to get the updated hypotheses out either.
One of the things this series has done for me is help me understand a little better just how complicated biological processes are. I don’t doubt that the alternate hypothesis will get refined over time, but it blows my mind that much of the health services community is stuck on “fatties eat too much. duh!” with no thought to the concept that (some of) our bodies might respond in fundamentally different ways to different macronutrients.
Yes, that is my concern, also, which is partly why I’m writing this post (not the lay press would read anything I write).
Peter, you and others have said the best heart disease indicator on a standard lipid panel is Trig/HDL. Would Trig/HDL-P be a better indicator? You showed that Niacin raises HDL-C but in a way that offers no added heart health protection. Does Niacin affect LDL-P? Does Metformin raise HDL-C/P or lower LDL-P? Thanks, Dave
It might be (I don’t know if it’s been validated) and the units don’t really make sense. The TG/HDL-C ratio “works” because both are units of mg/dL. The advantage of this ratio is “everyone” can get it. It’s cheap and easily accessible. Niacin does have some impact on LDL-P, but not much, and it depends on the dose and the underlying reason for elevated LDL-P.
You can not use lipid concentrations and lipoprotein concentration ratios in the same ratio – no relationship. Niacin significantly lowers apoB and LDL-P and that is it’s major benefit. It raises HDL-C by making HDL particles larger but does not raise total HDL-P. Thus it is not an effective HDL drug. Metformin can lower LDL-P.
Your cartoon story is fantastic
Dr Lipid
Not specifically about this post, but you have emphasized that LDL-P is the only significant number regarding cholesterol. My question is (maybe I’ve missed this), in the absence of inflammation, will the particles be able to penetrate the arterial wall? In other words, does LDL-P even matter if there’s no systemic inflammation? Or perhaps it depends on the degree of inflammation?
Typically inflammation is the result of the following sequence: LDL particle crosses endothelium, gets retained in S-E space while proteoglycans “hold” it in place, inflammatory cells arrive and begin immune response to modulate “invader.”
Hi Peter,
Thanks for the holy grail of cholesterol knowhow.
In regards to your saying: inflammation is a consequence of LDL-P crossing the endothelium. Then if i see the other way around, if there’s not much inflammation i would assert that not much of LDL-P particles have been stored in the endothelium right?
I must admit that I view this scenario as including two different phases of inflammation; the one that is caused by the retention of the particle in the S-E space, and the one that is systemic and attracts the particle to the endothelium in the first place. Apologies, but my physiologist’s gut still does not allow me to accept the concentration gradient causing “crash” idea. Of the various species in circulation at any given time, why is it only the LDL particle that crashes? It seems to me that what distinguishes LDL-p from everything else is ApoB, and in my scenario, ApoB is being signalled into the endothelium. My money says that ApoB has an immune function which is being signalled by an existing inflammatory process in the vessels and system wide, caused by autoimmunity and/or infection. There’s no crash at all in this scenario, but rather signalling. This idea has some consistency with observations of changes in cholesterol concentration with sepsis and I believe there was some work done on another Apo molecule on HDL as a potential mediator of this. I got 50 bucks on this if anyone wants to take the bet. Potential candidates as cause are autoimmunity/infection from intestinal permeability or injury from blood pressure.
You might be right, Lorraine. Let’s assume there is a specific immune trigger uniquely or at least “affectionately” directed towards apoB. What is a better treatment? Knock down the immune system? Or reduce apoB? In my former life, both in medical school and my fellowship, I worked specifically on immunology. The former problem is MUCH more difficult than the latter. In fact, we have many solutions to the latter, and very few to the former. The solutions we do have to the former come with side-effects that make anti-apoB treatments look like drinking water. Even if (like I said, this is a valid argument) we could put people of FK506 to reduce their inflammatory response to the apoB particles, what other mess have we created?
Ah, perhaps we should say that there are few if any agreeable *pharmaceutical* solutions to “knocking down the immmune system”. I wouldn’t suggest that approach at all. How about reducing the potential antigen load altogether by healing the permeable intestine? This is the basis of Robb Wolf’s entire perspective, as well as that of the Weston Price folks. And in the end, the solution comes back to what you’re doing, a diet that is as much anti-inflammatory as it is anything else. Many of the foods that we eliminate in carb restriction are the very suspects in autoimmunity.
I’m also not suggesting that we shouldn’t treat the known agents in atherosclerosis. My point of view is merely that the cascade of events described once the LDL particle retains is possibly secondary to a previous inflammatory event that causes that cascade.
Ah, I see your point. Yes, agree with you.
It just seemed to me that if there’s no event like inflammation to force the particles into the arterial wall that what you’re saying, in essence, is that we’re all doomed, some just faster than others, because this is a natural process that happens to everyone.
Yes, some would argue this. Since age is the single greatest risk factor for CVD it suggests that eventually everyone will succumb to this process, even if every other disease could be mitigated. A 10 year old with and LDL-P of 3,000 has a lower 5 year risk than a 70 year old with an LDL-P of 1,000. Why? Probably because the 70-year old’s arteries have had much longer aggregate exposure to apoB particles.
Lorraine, my money’s on you! 🙂 I put down 50 EUR sayin’ that the ApoB particles only (excessively!) find their way into the subendothelial space if they are signaled-in by pathogenic factors. TNF-alpha, anyone? 🙂
Peter, age is obviously a factor, but you can’t rule out exposure history to pathogens as the driving factor, over the exposure to a body-produced particle that is a critical part of metabolism. Is there any data toward this (pathogens vs. age)? I doubt it, it’s bound to be virtually impossible to measure.
Hi Peter.
You know, I’m re-reading GCBC, and the whole chapter about insulin and cardiovascular disease makes wonder how you could name your blog (initially) as “The War on Insulin” and then forget about insulin in this series of articles about cholesterol.
In fact, according to Taubes, the smoking gun of arterial wall disruption is most probably in the hands of insulin.
To me, it looks like it is the combination of active disruption (by insulin and blood sugars) AND the available by-standing ammunition (Apo-B containing lipoprotein particles) that is responsible for atherosclerosis.
Which begs the question (which I take it is being observed in people like me), that perhaps a high Apo-B particle count (high LDL-P) is NOT more atherosclerotic than a low LDL-P while in the absence of high insulin?…
This is a very good point, Vasco, and it is the heart of the argument/thesis put forth by folks like Gary Taubes, Eric Westman, and several others, as to why the standards of predictive cut-offs may not apply to the population you describe. However, until we do a properly designed clinical trial to test this hypothesis, it remains a hypothesis. It makes sense, I agree, but we only have bits of the story worked out. Remember the words of Thomas Henry Huxley (Darwin’s “bulldog”): “The great tragedy of science is the slaying of a beautiful hypothesis by an ugly fact.”
Peter – any comments on the famous HDL ratios that we are all supposed to be paying so much attention to? Would you say that it doesn’t matter at all, or only to a point? I would guess that given the limited correlation between LDL-P and LDL-C in “most” people, a “bad” ratio usually is bad but a “good” ratio may not mean much at all?
I certainly wouldn’t say they don’t matter. I like to think of the TG/HDL-C as a “poor man’s test” for IR. When someone has a ratio less than 1.0 (i.e., they have more HDL-C than TG), odds are they are doing well. That said, if I had a dollar for every time I saw a normal ratio there, but a stronger marker of IR elsewhere (e.g., HOMA-IR, LP-IR), I’d have a lot of dollars. There is no one number that is “perfect” and there is no test, even LDL-P, that should be taking in complete isolation.
The correlation between LDL-P and LDL-C is not at all “limited”….its just not perfect. (I will try to find time to dig out some correlation coefficients on this). Their high degree of correlation is why LDL-P has not been found to add predictive power to LDL-C.
As for ratios, in all the studies I looked at TC/HDL was superior to LDL-P as a predictor so hardly a “poor man’s” substitute there.
It’s about 0.7; very similar to that of HDL-P and HDL-C.
Well, .7 is considered “high.”
Also, I should have said “TC/HDL was EQUAL TO to LDL-P”, not necessarily superior.
Dear Peter,
I have an off topic question. I have been reading your blog nearly since it started. My 22 yr old son is home for the summer and so he has automatically gone on a low carb diet. He signed up with a gym and the trainer there told him to get off of low carb for weight training.
I have ordered Jeff Volek’s book Art and Science of Low Carb Performance. But, it seems like you addressed this issue but can’t find the post without rereading reams of material.
Check out the posts about the interplay of exercise and ketosis (2 parts). Of course, your son does not need to be in ketosis. Simply removing sugar and highly refined grains will do wonders for him.
Hi Pam:
Ok, so I got Volek’s book as soon as it came out, and got a blood ketone meter 1 week later.
As a woman, I was at 50 g total carbs a day, eating 65% fat, 25% protein, and 10% carbs for 1750 calories. I lift Superslow and at the time my max leg press was 305.
My husband was at 65g total carbs a day, eating 60% fat, 25% protein and 15% carbs for 2100 calories a day. He swims at Stanford 3x a week and played a game of water polo.
We started out at 1.4 and .4 on the ketone meter. After 1 week of implementing Volek’s suggestions, I switched to eating 75% fat, 18% protein, and the rest carbs, going down to 30 total g a day. My calories remained the same.
My husband made similar changes. His ketones shot up to 2.4 and his stroke rate improved (he swam the pool lengths with fewer strokes). He also dropped to 8.5% body fat, down from 10. He lost an inch off his waist and his chest muscles suddenly appeared to balloon.
The effect for him was really dramatic. My leg press weight, instead of going up 1/2 or 1 pound a week, went up 3 pounds to 308!
As time has gone on, we have continued to improve. My leg press after about 5-6 weeks of Volek’s advice is now 320. My husband’s swimming continues to improve – he swims more tirelessly and has greater mental focus in the moment, allowing him to really concentrate on improving the details of his Total Immersion technique.
He believed his balance in the water is much better and that he has more power in what is called “hip drive.”
Volek himself pushes 1/2-ton boulders and was a weight-lifting champion. He personally lives at 1.5-2.0 on the ketone meter. I can attest there are many athletic and mental benefits to this level of ketones.
@ Pam F
High-carb pre-workout, low-carb the rest of the time; high-protein post-workout (recovery’s where you make your gains), low pro the rest of the time. So, he will be high-fat, low-carb, low-pro outside the pre- and post-lifting windows.
The carbs will power the weight-lifting, making the session maximally productive/efficient; the lifting session will burn off those carbs.
(“High-carb doesn’t mean go hog wild, nor is it an excuse to eat junk food.)
https://www.jeffreybrauer.blogspot.com/2012/03/how-to-sync-diet-with-exercise.html
solid information
Great stuff once again. The shift in thinking about HDL-c is one of the more difficult things for people (including myself) to wrap their heads around. In trying to share this information with people, it’s difficult to get past their incredulity at the fact that just because their HDL-c is high doesn’t mean their arteries are teflon coated works of art that are impervious to insult. Hopefully there are a lot of clinicians reading what you and some of the other people on the forefront of this topic are writing.
Interestingly, in the (admittedly different) context of their LP-IR score, Liposcience’s green-red line puts small HDL size on the negative side.
Good point. Suggests how confusing this whole area is…
The only LipoSciene LP-IR score predicts is insulin resistance – not CV risk!!! It is also only valid in drug naive patients, since drugs (artificially) interfere with size and the score hasn’t been validated under these conditions.
DR Lipid
Is someone considered “drug-naive” if they are taking red yeast rice (CAM alternative to a statin)? And should RYR be avoided without monitoring sterol absorption and synthesis, since statin momotherapy is contraindicated in some people? Thanks, maryann
Hmmm. That’s a good question. I don’t know the answer. It depends on how much it impacts particle size and number.
Understood. But then small HDL size may be bad for IR but may be good for CV. Just saying that it will be interesting to see how this plays out.
Completely agree. The journey is as much fun as the destination.
Peter, I’m seeing that the NMR or test for IR may be inaccurate when one is taking cholesterol lowering meds. I’m in that boat & having my tests this week. Will I get anything beneficial from these tests under the circumstances? I’ve been 7 weeks Keto adapted but still not losing on scale.
Correct, the LP-IR has only been validated in patients not taking cholesterol-lowering drugs.
“you can’t have trials in medicine without catchy names!”
SO true! And it seems like the more they have to cleverly torture the language to come up with the “appropriate” acronym, the better they like it.
Peter, take a look at this paper by Dr. Asztalos, et al. entitled “High-Density Lipoprotein Subpopulation Profile and Coronary Heart Disease Prevalence in Male Participants of the Framingham Offspring Study.” It’s at https://atvb.ahajournals.org/content/24/11/2181.full.pdf+html
In this study, Dr. Aztalos used 2-dimensional Gradient Gel Electrophoresis to subdivide HDL into several different particle sizes and charges, from the smallest, pre-beta HDL particles to the largest particles, alpha-1. In this study he shows that the parameter that best correlates with with no CHD is a high concentration of alpha-1 particles, which are quite large as HDL particles go(>11nm). In the study reported in this reference, the prevalence of CHD was almost linear (and inverse) with alpha-1 HDL. Note that there were about 106 Framingham Offspring out of a total of 1446 with alpha-1 of 30 or greater, and not one of them had CHD. All of the 169 people with CHD had alpha-1 less than 30, and most had alpha-1 less than 20. Neither of the other two common measures of HDL, HDL-C or ApoA-1, are as clearly definitive about CHD risk as alpha-1, which has an almost monotonic curve of alpha-1 vs risk, as may be seen from Figure 2 of the paper. In short, the more alpha-1 the better. Which suggests that the more large HDL a person has, the less likely to develop CHD. This study prevents me from concluding that small HDL is more protective than large HDL.
Harry, this study has some limitations. HDL-P via NMR was not measured (they Boston heart lab uses 2 dimensional gel electrophoresis with apoA-I staining) and confirms that people at risk lack large HDL particles — although not measured in this study, the risk is likely due to high LDL-P and low total HDL-P due to catabolism of TG-rich large HDLs and delayed maturation of prebeta HDL (not captured by NMR).
Good stuff, but it made my brain work extra hard. Thanks for putting this info out, I need to re read this so I make sure I’m understanding everything. I’ve started writing some posts on cholesterol for my non-sciencey friends and family. I just hope I’m not totally botching it up as I try to simplify the concepts…because they really aren’t simple. I have learned so much from this series, anxiously awaiting the next one!
Last night wife, while reading this post, suggested I take a break from it for a while. She’s worried the blog is getting too “technical” and that I’m going to turn many folks off. I see her point. The cholesterol series isn’t exactly lunchroom discussion. I’m glad you find it interesting enough to turn it into family/friend discussions.
Dr. Attia, your wife may indeed have a point. As much as I have enjoyed reading the seven parts (and I was initially expecting a total of 3 parts, maybe) so far posted on this series and learning enough to make my endocrinologist look completely puzzled with the extent of my knowledge, I’m really searching for the answer to one question: what should I do to improve my odds of living a longer, healthier life? Honestly, I think we’re taking too long to get to it. I’m sure the answers, if at all known, will come with several qualifications about the precarious state of our knowledge. That’s fine. But, please, get to it soon…
I may start alternating “lighter” posts with cholesterol posts. I think there are still around 3 or 4 more cholesterol posts waiting to be written.
Oh,no! If you start alternating posts it will take further two months for us to get to the effects of diet, statins, exercise, etc. over CVD…
I like the “alternating” idea — especially since there are so many great topics on the wish list! I’d also consider some videos on the cholesterol content — that’d make the material more accessible for folks who prefer to learn that way.
Big picture though, this blog is fantastic. Thank you!
I vote for continuing the deep dive into cholesterol. As with others, I can’t wait for the payoff.
That being said, I did not mind the timely Bloomberg digression as it is getting even more nuts out there since, they are now proposing banning buttered popcorn and milkshakes. (It’s a short, slippery road to red meat and butter…).
Like JW, I’m confused here. I have always read that Large HDL-P is more protective than Small HDL particles. Are you saying that the new wisdom on HDL particles is itself mistaken?
It might be. I don’t think we can say definitively. The REAL challenge is understanding for certain the cause vs. the bystander. For example, we know people with small LDL particle tend to have more. But what is cause of their greater disease risk? Size or number or both? Jim Otvos’ amazing analysis which I reviewed previously, suggests size is NOT an independent predictor of risk, so it’s all particle number. We have less data on the HDL size question, and functionality of the particles plays a very important role. As I said, we can’t “measure” this, so we’re stuck a bit.
HDL size per se has no clinically important meaning and the NLA discourages its measurement. HDL particles undergo a dynamic remodeling process (lipidation followed by delipidation) No tests are available to measure this. If you had to pick a size, most experts would say the small HDL is the most crucial.
Hope my questions r.e. men vs women aren’t getting tiresome- but I was wondering if the graph of the Framingham data showing that small HDL particles increase up to about 40 then go down is from men only, or pooled men and women. (I assume it isn’t from women only.) I ask this because my understanding is that the good/optimal levels of HDL-C are defined as higher for women than men. I was wondering if this correlates with a higher level of total HDL-C at which the increase in small HDL particles stops in women as compared to men.
Thanks
Laura, how could I find a question tiresome that impacts 50% of the population? If you were asking questions about redheads, who were born on the eve of the summer solstice on leap years, who are between 6-foot-10.5 and 6-foot-11, who had purple eyes, I might find it tiresome.
Those data are from men and women.
Peter- I am completely engrossed in this series. Thanks for you efforts. Perhaps you will address this in a future post (if so, I can patiently wait), but I’ve heard conflicting statements regarding whether there have been any studies that definitively show that cholesterol lowering medications reduce mortality in patients who have not yet had a cardiac event. I have had one physician tell me that obviously drug companies would love nothing more to be able to demonstrate this, yet they have not been able to do so. I am curious as to which studies, if any, show that lowering cholesterol through medication lowers cardiovascular risk or mortality rates in patients that haven’t had a cardiac event.
Thanks again,
Mike
Mike, this is a very important topic, and one that warrants an entire post — which it will receive. What you’re asking about is called “primary prevention,” and there is a lot to discuss.
I read the book “The great cholesterol con” and one of the authors principal conclusions was that Statins didn’t help women at all, and only helped men who had already had a heart incident. After reading Peter’s and others work, I think there might be a more nuanced answer here but in general there is not a lot of data supporting statins for patients without a previous heart incident. Once you factor in LDL-P in to the equation I think statins can play a role, but that data wasn’t available for the author of that book. I believe in general that statins are over prescribed and that their use needs to be to treat the correct symptom (i.e. non LDL-C).
I agree completely (despite my back and forth with one particularly erudite reader) — statins are almost certainly over-prescribed. But the binary view (“Everyone should be on statins” versus “Statins are horrible and should be abolished”) is utterly baseless and naive. Statins are a tool. Simple and plain. Use the tool for the RIGHT job at the RIGHT time, and it helps. If you don’t, it doesn’t, and it might cause harm.
Low risk people do not die and thus mortality cannot be tested unless you study 50000 folks for 30 years. No one will pay for such a study, of course.
I was going to comment on last weeks ping pong match with said erudite reader but decided that the thread had run its course. I think in general he felt that any preventative testing would be followed up by drugs to treat. In some larger sense he has a point that there are a lot of diagnostic tests out there and if you test long and hard enough you’ll probably find something. In the pragmatic world we live in I think there is some filtering by physicians as to the tests they think their patients need based on gender, age, race etc. I don’t think my 8 year old daughter needs an NMR lipid panel (even though she lives on ice cream and waffles). I on the other hand got this and a few other selective tests done. It’s all probability & statistics and common sense.
On another point, it would appear that the drug manufacturers have thrown a lot of money at the raise HDL-C hypotheis and have not had anything stick. You can imagine the profit potential for those companies if they found a drug that actually improved CVD without side effects. I would imagine that they are revising their hypothesis about HDL-C about now. It would seem that forcing the body to make more HLD-C upsets the balance of the system and that there is a different mechanism in play when drugs are in play versus someone who has naturally occuring high HDL-C. Nature does not give up its secrets easily it would seem. Keep up the great work.
Yes, I think you’re taking the right approach, David.
Regarding statins and all-cause mortality.: As Dr. Dayspring points out, it’s a very long-term and expensive project to do a mortality study on low risk (primary prevention) folks. Of course, heart disease risk correlates best with age, so to get a higher risk test group you need to test old people. One key thing that is almost completely overlooked is that, in people over age 60 or 65, higher cholesterol is associated with reduced mortality rate (lots of studies show this). Lower may well not be better for older folks, at least at some cut point (I would argue that guidelines-sanctioned levels may be relatively dangerous for the elderly). Just to be clear, I am throwing this out as hypothesis and not established fact.
We may not know whether statins provide their benefit (reduced heart attacks, but more reduction of the non-fatal variety and with 65% to 85% residual risk) by reducing LDL-P or through anti-inflammatory effects or something else, but they certainly do reduce cholesterol, and lower cholesterol in the elderly may be disadvantageous for longevity. This may not be the case for a middle-aged man with familial hypercholesterolemia and a family history of early heart disease. It may not be the case for other people at high enough risk. So, results may be mixed for different groups. At the moment, the received wisdom on prescribing statins makes little distinction regarding age or gender, and the presumption is that, once started, one should take statins for life.
I don’t take Dr. Dayspring‘s comment to mean that, if only it were practical to do a very long-term mortality study on statins in low-risk folks, we would certainly see a mortality rate benefit. Maybe we would or maybe we would see the opposite. Statins are very powerful drugs and are blunt instruments, interfering with a very significant biological process at an upstream point in order to lower blood lipid levels. Long-term bad effects might start to show up if there were seriously long-term trials (I quit Lipitor after 11 years due to side effects). Of course, long-term testing means that the cohort gets older, too, which (I hypothesize) makes it less likely that there would be a mortality rate benefit for statins.
In Uffe Ravnskoff’s book “The Cholesterol Myths” there’s an interesting sidelight on this (or a similar) niacin trial; there was no benefit while it lasted, but five years after it stopped there was a significant drop in CVD mortality in the niacin arm…
From memory I believe rodents lack CEPT and this is one of many differences in their lipid transport systems that make rats poor surrogates for humans in diet trials.
Hmmm. The niacin trial only stopped 2 years ago. I wonder if this is a different trial.
Peter,
I have a question about concordance/discordance as it relates to LDL-P and LDL-C which you may or may not be able to answer. If LDL-P and LDL-C are concordant while eating a ketogenic diet, will they typically or necessarly be in concordance while eating a traditional diet? A follow up question would be if they are also concordant while taking cholesterol lowering medication. In other words, is LDL-P and LDL-C concordance/discordance (not actual values but rather the correlation) influenced by exogenous factors? This may not be within the scope of your research and I agree with what you have stated before that there is further research that needed regarding how a ketogenic diet influences atherosclerosis beyond the known mechanicms – increased LDL-P, insulin resistance, infalmmatory markers, etc. Thanks for your time.
Great questions, Eric. I do not know the answer this, beyond some anecdotal observations (e.g., in myself – I went from being discordant to concordant). Obviously no conclusions can be drawn, as this has not been studied to my knowledge.
Another great installment to a fascinating series! I teach a class on chronic illness prevention through diet and lifestyle, and am hoping to incorporate your information before my next lecture. Please please please don’t stop until you cover the factors that influence particle number and size, and how to fix it when things go wrong.
Will do.
Yes seriously Dr. Attia, next Tuesday I’ll be the exact age my father was when he dropped dead of a heart attack. So, kind of a sense of urgency. If I suddenly quit leaving comments, you’ll know what happened…
Kevin, LOL! But Peter, I’m in the camp that deeply appreciates all the advanced learning that you are offering in this series and hope that you will proceed until your heart is content that you’ve delivered what you set out to accomplish with this information. You do a great job of explaining difficult and complex topics in a manner which is accessible to common sense, even if it takes a couple of reads for folks to get the lingo. And it has been amply demonstrated in the comments that a lot of your readers have real skin in the game in understanding this topic for their own well-being. Creating a sophisticated medical consumer is something I’m sure is a primary goal of NuSI.
Peter,
I am one of your readers who looks forward to the weekly post, and I sure hope you continue. However, if you and your wife do decide to take a break from the Cholesterol series, please don’t leave us with a cliff hanger until next season for some key questions — please at least give us a preview to some questions, such as:
(1) Once we get our LDL-P from Health Diagnostics Labs (HDL=cute), can we work towards modifying it through diet? And which foods, macronutrients, or nutrients lower the number — and which raise the number?
(2) You’ve said statins are wonderful tools, but not the best tool (like a hammer) for all situations (like cleaning windows). For which situations are statins in fact wonderful tools?
Many thanks!
Have no fear, Dorian. We aren’t done yet and certainly won’t be until I address these very important issues.