Want to catch up with other articles from this series?
- The straight dope on cholesterol – Part I
- The straight dope on cholesterol – Part II
- The straight dope on cholesterol – Part III
- The straight dope on cholesterol – Part IV
- The straight dope on cholesterol – Part V
- The straight dope on cholesterol – Part VI
- The straight dope on cholesterol – Part VII
- The straight dope on cholesterol – Part VIII
- The straight dope on cholesterol – Part IX
Previously, in Part I, Part II, Part III, Part IV, Part V ,and Part VI of this series, we addressed these 8 concepts:
#1 — What is cholesterol?
#2 — What is the relationship between the cholesterol we eat and the cholesterol in our body?
#3 — Is cholesterol bad?
#4 — How does cholesterol move around our body?
#5 – How do we measure cholesterol?
#6 – How does cholesterol actually cause problems?
#7 – Does the size of an LDL particle matter?
#8 – Why is it necessary to measure LDL-P, instead of just LDL-C?
(No so) Quick refresher on take-away points from previous posts, should you need it:
- Cholesterol is “just” another fancy organic molecule in our body but with an interesting distinction: we eat it, we make it, we store it, and we excrete it – all in different amounts.
- The pool of cholesterol in our body is essential for life. No cholesterol = no life.
- Cholesterol exists in 2 forms – unesterified or “free” (UC) and esterified (CE) – and the form determines if we can absorb it or not, or store it or not (among other things).
- Much of the cholesterol we eat is in the form of CE. It is not absorbed and is excreted by our gut (i.e., leaves our body in stool). The reason this occurs is that CE not only has to be de-esterified, but it competes for absorption with the vastly larger amounts of UC supplied by the biliary route.
- Re-absorption of the cholesterol we synthesize in our body (i.e., endogenous produced cholesterol) is the dominant source of the cholesterol in our body. That is, most of the cholesterol in our body was made by our body.
- The process of regulating cholesterol is very complex and multifaceted with multiple layers of control. I’ve only touched on the absorption side, but the synthesis side is also complex and highly regulated. You will discover that synthesis and absorption are very interrelated.
- Eating cholesterol has very little impact on the cholesterol levels in your body. This is a fact, not my opinion. Anyone who tells you different is, at best, ignorant of this topic. At worst, they are a deliberate charlatan. Years ago the Canadian Guidelines removed the limitation of dietary cholesterol. The rest of the world, especially the United States, needs to catch up. To see an important reference on this topic, please look here.
- Cholesterol and triglycerides are not soluble in plasma (i.e., they can’t dissolve in water) and are therefore said to be hydrophobic.
- To be carried anywhere in our body, say from your liver to your coronary artery, they need to be carried by a special protein-wrapped transport vessel called a lipoprotein.
- As these “ships” called lipoproteins leave the liver they undergo a process of maturation where they shed much of their triglyceride “cargo” in the form of free fatty acid, and doing so makes them smaller and richer in cholesterol.
- Special proteins, apoproteins, play an important role in moving lipoproteins around the body and facilitating their interactions with other cells. The most important of these are the apoB class, residing on VLDL, IDL, and LDL particles, and the apoA-I class, residing for the most part on the HDL particles.
- Cholesterol transport in plasma occurs in both directions, from the liver and small intestine towards the periphery and back to the liver and small intestine (the “gut”).
- The major function of the apoB-containing particles is to traffic energy (triglycerides) to muscles and phospholipids to all cells. Their cholesterol is trafficked back to the liver. The apoA-I containing particles traffic cholesterol to steroidogenic tissues, adipocytes (a storage organ for cholesterol ester) and ultimately back to the liver, gut, or steroidogenic tissue.
- All lipoproteins are part of the human lipid transportation system and work harmoniously together to efficiently traffic lipids. As you are probably starting to appreciate, the trafficking pattern is highly complex and the lipoproteins constantly exchange their core and surface lipids.
- The measurement of cholesterol has undergone a dramatic evolution over the past 70 years with technology at the heart of the advance.
- Currently, most people in the United States (and the world for that matter) undergo a “standard” lipid panel, which only directly measures TC, TG, and HDL-C. LDL-C is measured or most often estimated.
- More advanced cholesterol measuring tests do exist to directly measure LDL-C (though none are standardized), along with the cholesterol content of other lipoproteins (e.g., VLDL, IDL) or lipoprotein subparticles.
- The most frequently used and guideline-recommended test that can count the number of LDL particles is either apolipoprotein B or LDL-P NMR, which is part of the NMR LipoProfile. NMR can also measure the size of LDL and other lipoprotein particles, which is valuable for predicting insulin resistance in drug naïve patients, before changes are noted in glucose or insulin levels.
- The progression from a completely normal artery to a “clogged” or atherosclerotic one follows a very clear path: an apoB containing particle gets past the endothelial layer into the subendothelial space, the particle and its cholesterol content is retained, immune cells arrive, an inflammatory response ensues “fixing” the apoB containing particles in place AND making more space for more of them.
- While inflammation plays a key role in this process, it’s the penetration of the endothelium and retention within the endothelium that drive the process.
- The most common apoB containing lipoprotein in this process is certainly the LDL particle. However, Lp(a) and apoB containing lipoproteins play a role also, especially in the insulin resistant person.
- If you want to stop atherosclerosis, you must lower the LDL particle number. Period.
- At first glance it would seem that patients with smaller LDL particles are at greater risk for atherosclerosis than patients with large LDL particles, all things equal.
- “A particle is a particle is a particle.” If you don’t know the number, you don’t know the risk.
- With respect to laboratory medicine, two markers that have a high correlation with a given outcome are concordant – they equally predict the same outcome. However, when the two tests do not correlate with each other they are said to be discordant.
- LDL-P (or apoB) is the best predictor of adverse cardiac events, which has been documented repeatedly in every major cardiovascular risk study.
- LDL-C is only a good predictor of adverse cardiac events when it is concordant with LDL-P; otherwise it is a poor predictor of risk.
- There is no way of determining which individual patient may have discordant LDL-C and LDL-P without measuring both markers.
- Discordance between LDL-C and LDL-P is even greater in populations with metabolic syndrome, including patients with diabetes. Given the ubiquity of these conditions in the U.S. population, and the special risk such patients carry for cardiovascular disease, it is difficult to justify use of LDL-C, HDL-C, and TG alone for risk stratification in all but the most select patients.
- To address this question, however, one must look at changes in cardiovascular events or direct markers of atherosclerosis (e.g., IMT) while holding LDL-P constant and then again holding LDL size constant. Only when you do this can you see that the relationship between size and event vanishes. The only thing that matters is the number of LDL particles – large, small, or mixed.
Concept #9 – Does “HDL” matter after all?
Last week was the largest annual meeting of the National Lipid Association (NLA) in Phoenix, AZ. The timing of the meeting could not have been better, given the huge buzz going around on the topic of “HDL.” (If you’re wondering why I’m putting HDL in quotes, I’ll address it shortly.)
What buzz, you ask? Many folks, including our beloved health columnists at The New York Times, are talking about the death of the HDL hypothesis – namely, the notion that HDL is the “good cholesterol.”
Technically, this “buzz” started about 6 years ago when Pfizer made headlines with a drug in their pipeline called torcetrapib. Torcetrapib was one of the most eagerly anticipated drugs ever, certainly in my lifetime, as it had been shown to significantly raise plasma levels of HDL-C. You’ll recall from part II of this series, HDL particles play an important role in carrying cholesterol from the subendothelial space back to the liver via a process called reverse cholesterol transport (RCT). Furthermore, many studies and epidemiologic analyses have shown that people with high plasma levels of HDL-C have a lower incidence of coronary artery disease.
In the case of torcetrapib, there was an even more compelling reason to be optimistic. Torcetrapib blocked the protein cholesterylester transfer protein, or CETP, which facilitates the collection and one-to-one exchange of triglycerides and cholesterol esters between lipoproteins. Most (but not all) people with a mutation or dysfunction of this protein were known to have high levels of HDL-C and lower risk of heart disease. Optimism was very high that a drug like torcetrapib, which could mimic this effect and create a state of more HDL-C and less LDL-C, would be the biggest blockbuster drug ever.
The past month or so has seen this discussion intensify, which I’ll quickly try to cover below.
The data
Torcetrapib
After several smaller clinical trials showed that patients taking torcetrapib experienced both an increase in HDL-C and a reduction in LDL-C, a large clinical trial pitting atorvastatin (Lipitor) against atorvastatin + torcetrapib was underway. This trial was to be the jewel in the crown of Pfizer. It was already known that Lipitor reduced coronary artery disease (and reduced LDL-C, though this may have been a bystander effect and real reduction in mortality may be better attributed to the reduction in LDL-P).
I still remember exactly where I was standing, on the corner of Kerney St. and California St. in the heart of San Francisco’s financial district, on that December day back in 2006 when it was announced the trial had been halted because of increased mortality in the group receiving torcetrapib. In other words, adding torcetrapib actually made things worse. I was shocked.
Many reasons were offered for this, including the notion that torcetrapib was, indeed, helpful, but because of unanticipated side-effects, (raising blood pressure in some patients and altering electrolyte balance in others), the net impact was harmful. Some even suggested that the drug could be useful in the “right” patients (e.g., those with low HDL-C, but normal blood pressure). Furthermore, in two subsequent studies looking at carotid IMT (thickening of the carotid arteries) and intravascular ultrasound, there was no reduction in atherosclerosis.
This was a big strike against the HDL hypothesis and work on torcetrapib was immediately halted.
Niacin
Niacin has long been known to raise HDL-C and has actually been used therapeutically for this reason for many years. The AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides – you can’t have trials in medicine without catchy names!) sought to test this. The trial randomly assigned over 3,000 patients with known and persistent, but stable and well treated cardiovascular risk, to one of two treatments:
- Simvastatin (40-80 mg/day), +/- ezetimibe (10 mg/day) as necessary to maintain LDL-C below 70 mg/dL + placebo (a tiny dose of crystalline niacin to cause flushing);
- As above, but instead of a placebo, patients were given 1,500 to 2,000 mg/day of extended-release niacin.
Both arms of the study had their LDL-C < 70 mg/dL, non-HDL-C < 100 md/dL and apoB < 80 mg/dL, but despite the statin or statin + ezetimibe treatment still had low HDL-C. So, if niacin raised HDL-C and reduced events, the HDL raising hypothesis would be proven.
Simvastatin, as its name suggests, is a statin which primarily works by blocking HMG-CoA reductuse, an enzyme necessary to synthesize endogenous cholesterol. Ezetimibe works on the other end of problem, by blocking the NPC1L1 transporter on gut enterocytes and hepatocytes at the hepatobiliary junction (for a quick refresher, go back to part I of this series and look at the second figure – ezetimibe blocks the “ticket taker” in the bar).
After two years the niacin group, as expected, had experienced a significant increase in plasma HDL-C (along with some other benefits like a greater reduction in plasma triglycerides). However, there was no improvement in patient survival. The trial was futile and the data and safety board halted the trial. In other words, for patients with cardiac risk and LDL-C levels at goal with medication niacin, despite raising HDL-C and lowering TG, did nothing to improve survival. This was another strike against the HDL hypothesis.
Dalcetrapib
By 2008, as the AIM-HIGH trial was well under way, another pharma giant, Roche, was well into clinical trials with another drug that blocked CETP. This drug, a cousin of torcetrapib called dalcetrapib, albeit a weaker CETP-inhibitor, appeared to do all the “right” stuff (i.e., it increased HDL-C) without the “wrong” stuff (i.e., it did not appear to adversely affect blood pressure). It did nothing to LDL-C or apoB.
This study, called dal-OUTCOMES, was similar to the other trials in that patients were randomized to either standard of care plus placebo or standard of care plus escalating doses of dalcetrapib. A report of smaller safety studies (called dal-Vessel and Dal-Plaque) was published a few months ago in the American Heart Journal, and shortly after Roche halted the phase 3 clinical trial. Once again, patients on the treatment arm did experience a significant increase in HDL-C, but failed to appreciate any clinical benefit. Another futile trial.
Currently, two additional CETP inhibitors, evacetrapib (manufactured by Lilly) and anacetrapib (manufactured by Merck) are being evaluated. They are much more potent CETP inhibitors and, unlike dalcetrapib, also reduce apoB and LDL-C and Lp(a). Both Lilly and Merck are very optimistic that their variants will be successful where Pfizer’s and Roche’s were not, for a number of reasons including greater anti-CETP potency.
Nevertheless, this was yet another strike against the HDL hypothesis because the drug only raised HDL-C and did nothing to apoB. If simply raising HDL-C without attacking apoB is a viable therapeutic strategy, the trial should have worked. We have been told for years (by erroneous extrapolation from epidemiologic data) that a 1% rise in HDL-C would translate into a 3% reduction in coronary artery disease. These trials would suggest otherwise.
Mendelian randomization
On May 17 of this year a large group in Europe (hence the spelling of randomization) published a paper in The Lancet, titled, “Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study.” Mendelian randomization, as its name sort of suggests, is a method of using known genetic differences in large populations to try to “sort out” large pools of epidemiologic data.
In the case of this study, pooled data from tens of studies where patients were known to have myocardial infarction (heart attacks) were mapped against known genetic alterations called SNPs (single nucleotide polymorphisms, pronounced “snips”). I’m not going to go into detail about the methodology because it would take 3 more blog posts., But, the reason for doing this analysis was to ferret out if having a high HDL-C was (only) correlated with better cardiovascular outcome, which has been the classic teaching, or if there was any causal relationship. In other words, does having a high HDL-C cause you to have a lower risk of heart disease or is it a marker for something else?
This study found, consistent with the trials I’ve discussed above, that any genetic polymorphism that seems to raise HDL-C does not seem to protect from heart disease. That is, patients with higher HDL-C due to a known genetic alteration did not seem to have protection from heart disease as a result of that gene. This suggests that people with high or low HDL-C who get coronary artery disease may well have something else at play.
Oh boy. This seems like the last nail in the casket of the entire “HDL” hypothesis, as evidenced by all of the front page stories worldwide.
The rub: the difference between HDL-C and HDL-P
The reason I’ve been referring to high density lipoprotein as “HDL,” unless specifically referring to HDL-C, is that HDL-P and HDL-C are not the same thing. Just as you are now intimately familiar with the notion that LDL-C and LDL-P are not the same thing, the same is true for “HDL” which simply stands for high density lipoprotein, and like LDL is not a lab assay. In fact, unpublished data from the MESA trial found that the correlation between HDL-C and HDL-P was only 0.73, which is far from “good enough” to say HDL-C is a perfect proxy for HDL-P.
HDL-C, measured in mg/dL (or mmol/L outside of the U.S.), is the mass of cholesterol carried by HDL particles in a specified volume (typically measured as X mg of cholesterol per dL of plasma). HDL-P is something entirely different. It’s the number of HDL particles (minus unlipidated apoA-I and prebeta-HDLs: at most 5% of HDL particles) contained in a specified volume (typically measured as Y micromole of particles per liter).
As you can see in the figure below (courtesy of Jim Otvos’ presentation at the NLA meeting 2 weeks ago), the larger an HDL particle, the more cholesterol it carries. So, an equal number of large versus small HDL particles (equal HDL-P) can carry very different amounts of cholesterol (different HDL-C). Of course, it’s never this simple because HDL particles, like their LDL counterparts, don’t just carry cholesterol. They carry triglycerides, too. Keep in mind, HDL core CE/TG ratio is about 10:1 or greater – if the large HDL carries TG, it will not be carrying very much cholesterol.

So, the important point is that HDL-C is not the same as HDL-P (which is also not the same as apoAI, as HDL particles can carry more than one apoAI).
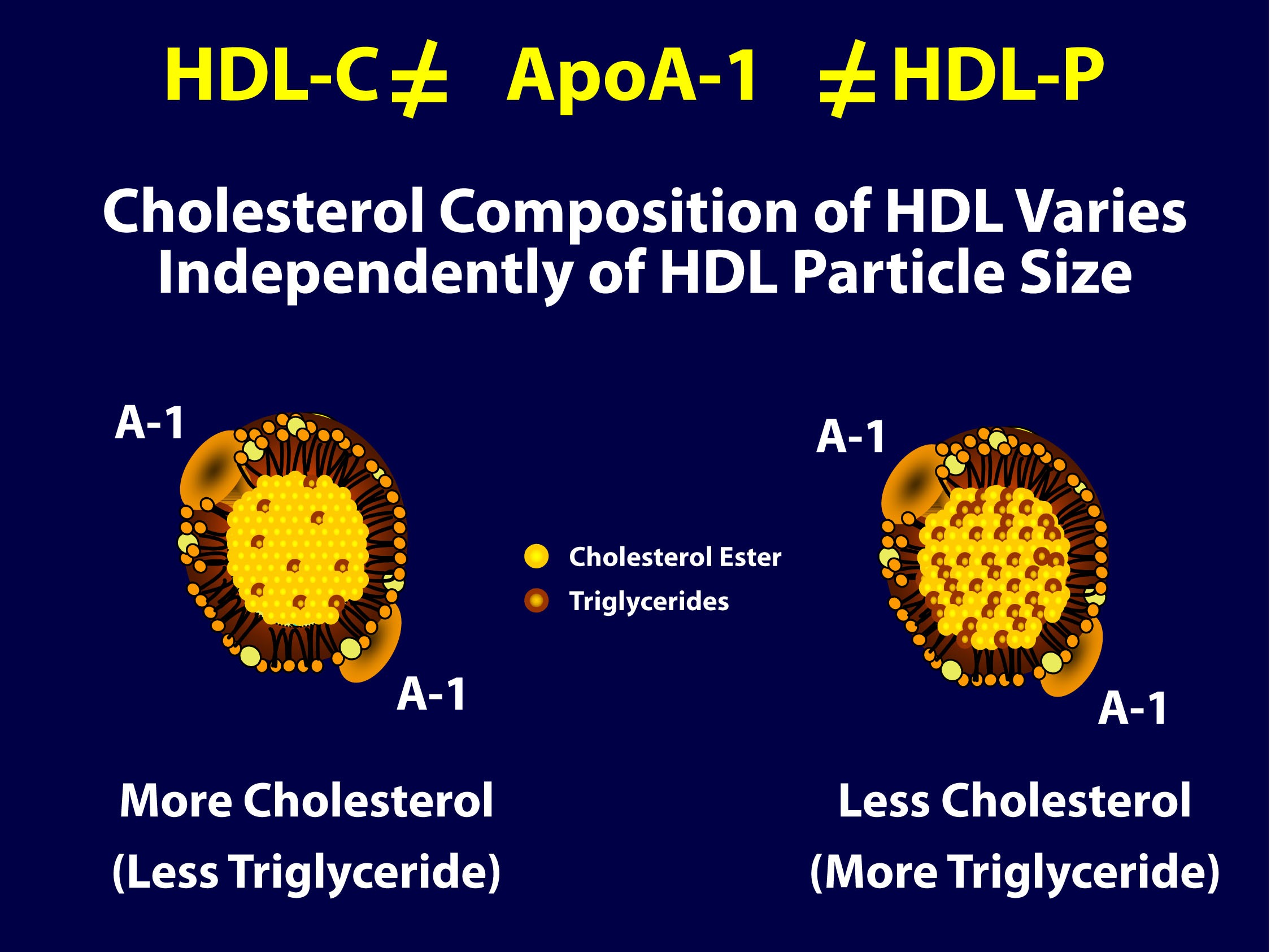
But there’s something else going on here. If you look at the figure below, from the Framingham cohort, you’ll note something interesting. As HDL-C rises, it does so not in a uniform or “across the board” fashion. A rise in HDL-C seems to disproportionately result from an increase in large HDL particles. In other words, as HDL-C rises, it doesn’t necessarily mean HDL-P is rising at all, and certainly not as much.
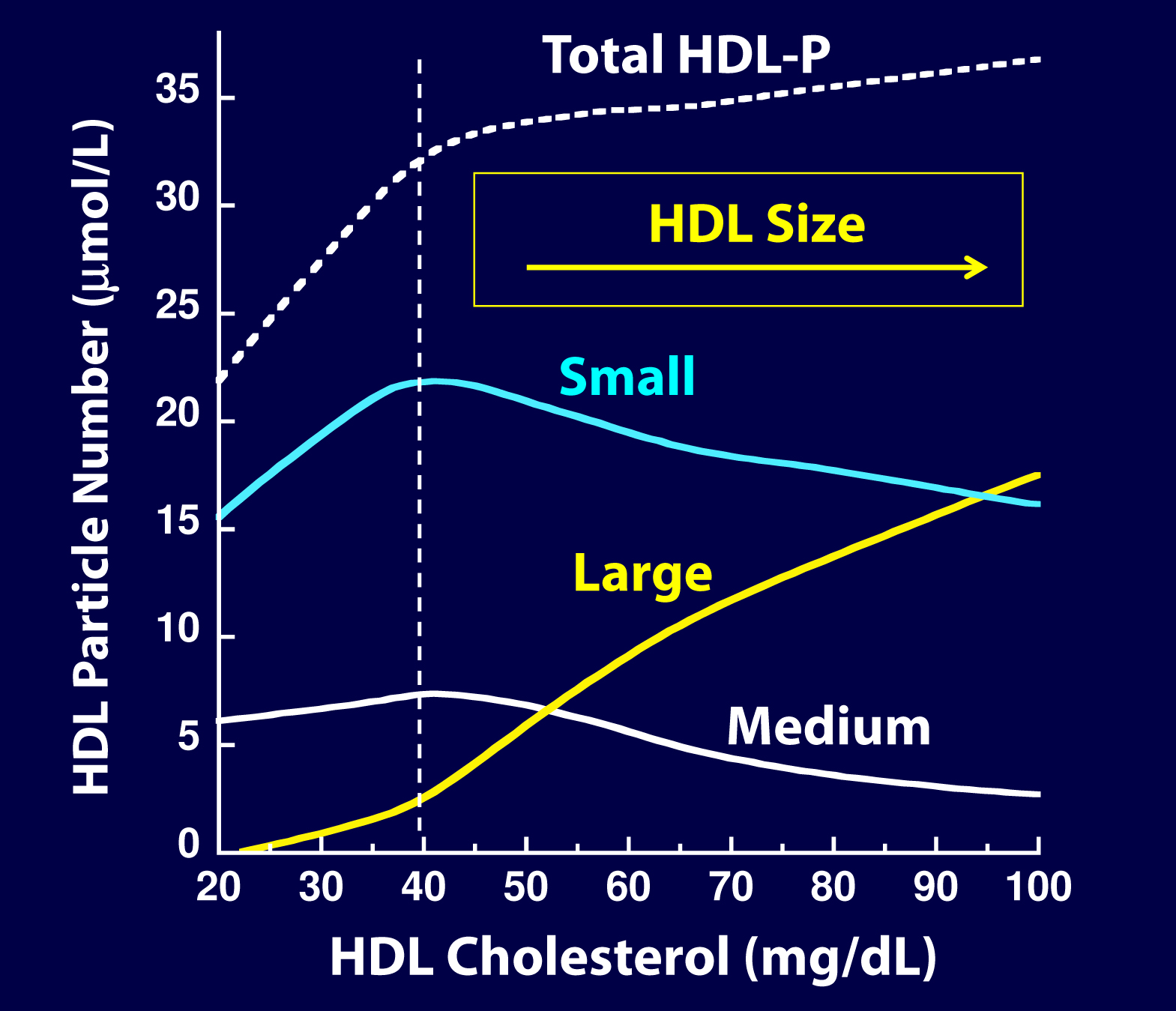
As you can see, for increases in HDL-C at low levels (i.e., below 40 mg/dL) the increase in small particles seems to account for much of the increase in total HDL-P, While for increases over 40 mg/dL, the increase in large particles seems to account for the increase in HDL-C. Also note that as HDL-C rises above 45 mg/dL, there is almost no further increase in total HDL-P – the rise in HDL-C is driven by enlargement of the HDL particle – more cholesterol per particle – not the drop in small HDL-P. This reveals to us that the small HDL particles are being lipidated.
Is there a reason to favor small HDL particles over large ones?
In the 2011 article, “Biological activities of HDL subpopulations and their relevance to cardiovascular disease,” published in Trends in Molecular Medicine, the authors describe in great detail some of protective mechanisms imparted by HDL particles.
Large HDL particles may be less protective and even dysfunctional in certain pathological states, whereas small to medium-sized HDL particles seem to confer greater protection through the following mechanisms:
- Greater antioxidant activity
- Greater anti-inflammatory activity
- Greater cholesterol efflux capacity
- Greater anti-thrombotic properties
In other words, particle for particle, it seems a small HDL particle may be better at transporting cholesterol from the subendothelial space (technically, they acquire cholesterol from cholesterol-laden macrophages or foam cells in the subedothelial space) elsewhere, better at reducing inflammation, better at preventing clotting, and better at mitigating the problems caused by oxidative free radicals.
Of course, reality is complicated. If there was no maturation from small to large HDL particles (i.e., the dynamic remodeling of HDL), the system would be faulty. So, the truth is that all HDL sizes are required and that HDL particles are in a constant dynamic state (or “flux”) of lipidating and delipidating, and the real truth is no particular HDL size can be said to be the best. If the little HDLs do not enlarge, the ApoA-I mediated lipid trafficking system is broken.
The truth about the old (and overly simplistic) term called reverse cholesterol transport (RCT)
HDL particles traffic cholesterol and proteins and last in plasma on average for 5 days. They are in a constant state of acquiring cholesterol (lipidation) and delivering cholesterol (delipidation). There are membrane receptors on cells that can export cholesterol to HDL particles (sterol efflux transporters) or extract cholesterol or cholesterol ester from HDL particles (sterol influx transporters).
The vast majority of lipidation occurs (in order): 1) at the liver, 2) the small intestine, 3) adipocytes and 4) peripheral cells, including plaque if present. The liver and intestine account for 95% of this process. The amount of cholesterol pulled out of arteries (called macrophage reverse cholesterol transport) is critical to disease prevention but is so small it has no effect on serum HDL levels. Even in patients with extensive plaque, the cholesterol in that plaque is about 0.5% of total body cholesterol. HDL particles circulate for several days as a ready reserve of cholesterol: almost no cell in humans require a delivery of cholesterol as cells synthesize all they need. However, steroidogenic hormone producing tissues (e.g., adrenal cortex and gonads) do require cholesterol and the HDL particle is the primary delivery truck.
If, as is the case in a medical emergency, the adrenal gland must rapidly make a lot of cortisone, the HDL particles are there with the needed cholesterol. This explains the low HDL-C typically seen in patients with severe infections (e.g., sepsis) and severe inflammatory conditions (e.g., Rheumatoid Arthritis).
Sooner or later HDL particles must be delipidated, and this takes place at: 1) the adrenal cortex or gonads 2) the liver, 3) adipocytes, 4) the small intestine (TICE or transintestinal cholesterol efflux) or give its cholesterol to an apoB particle (90% of which are LDLs) to return to the liver. A HDL particle delivering cholesterol to the liver or intestine is called direct reverse cholesterol transport (RCT), whereas a HDL particle transferring its cholesterol to an apoB particle which returns it to the liver is indirect RCT. Hence, total RCT = direct RCT + indirect RCT.
The punch line: a serum HDL-C level has no known relationship to this complex process of RCT. The last thing a HDL does is lose its cholesterol. The old concept that a drug or lifestyle that raises HDL-C is improving the RCT process is wrong; it may or may not be affecting that dynamic process. Instead of calling this RCT, it would be more appropriately called apoA-I trafficking of cholesterol.
Why do drugs that specifically raise HDL-C seem to be of little value?
As I’ve argued before, while statins are efficacious at preventing heart disease, it’s sort of by “luck” as far as most prescribing physicians are concerned. Most doctors use cholesterol lowing medication to lower LDL-C, not LDL-P. Since there is an overlap (i.e., since the levels of LDL-P and LDL-C are concordant) in many patients, this misplaced use of statins seems to work “ok.” I, and many others far more knowledgeable, would argue that if statins and other drugs were used to lower LDL-P (and apoB), instead of LDL-C, their efficacy would be even greater. The same is true for dietary intervention.
Interestingly, (and I would have never known this had Jim Otvos not graciously spent a hour on the phone with me two weeks ago giving me a nuanced HDL tutorial), a study that went completely unnoticed by the press in 2010, published in Circulation, actually did a similar analysis to the Lancet paper, except that the authors looked at HDL-P instead of HDL-C as the biomarker and looked at the impact of phospholipid transfer protein (PLTP) on HDL metabolism. In this study, though not the explicit goal, the authors found that an increase in the number of HDL particles and smaller HDL particles decreased the risk of cardiovascular disease. The key point, of course, is that the total number of HDL particles rose, and it was driven by increased small HDL-P. The exact same thing was seen in the VA-HIT trial: the cardiovascular benefit of the treatment (fibrate) was related to the rise in total HDL-P which was driven by the fibrates’ ability to raise small HDL-P.
It seems the problem with the “HDL hypothesis” is that it’s using the wrong marker of HDL. By looking at HDL-C instead of HDL-P, these investigators may have missed the point. Just like LDL, it’s all about the particles.
Summary
- HDL-C and HDL-P are not measuring the same thing, just as LDL-C and LDL-P are not.
- Secondary to the total HDL-P, all things equal it seems smaller HDL particles are more protective than large ones.
- As HDL-C levels rise, most often it is driven by a disproportionate rise in HDL size, not HDL-P.
- In the trials which were designed to prove that a drug that raised HDL-C would provide a reduction in cardiovascular events, no benefit occurred: estrogen studies (HERS, WHI), fibrate studies (FIELD, ACCORD), niacin studies, and CETP inhibition studies (dalcetrapib and torcetrapib). But, this says nothing of what happens when you raise HDL-P.
- Don’t believe the hype: HDL is important, and more HDL particles are better than few. But, raising HDL-C with a drug isn’t going to fix the problem. Making this even more complex is that HDL functionality is likely as important, or even more important, than HDL-P, but no such tests exist to “measure” this.