
Want to catch up with other articles from this series?
- The straight dope on cholesterol – Part I
- The straight dope on cholesterol – Part II
- The straight dope on cholesterol – Part III
- The straight dope on cholesterol – Part IV
- The straight dope on cholesterol – Part V
- The straight dope on cholesterol – Part VI
- The straight dope on cholesterol – Part VII
- The straight dope on cholesterol – Part VIII
- The straight dope on cholesterol – Part IX
Previously, across 8 parts of this series we’ve laid the groundwork to ask perhaps the most important question of all:
What should you eat to have the greatest chance of delaying the arrival of cardiovascular disease?
Before we get there, since this series has been longer and more detailed than any of us may have wanted, it is probably worth reviewing the summary points from the previous posts in this series (or you can just skip this and jump to the meat of this post).
What we’ve learned so far
- Cholesterol is “just” another fancy organic molecule in our body but with an interesting distinction: we eat it, we make it, we store it, and we excrete it – all in different amounts.
- The pool of cholesterol in our body is essential for life. No cholesterol = no life.
- Cholesterol exists in 2 forms – unesterified or “free” (UC) and esterified (CE) – and the form determines if we can absorb it or not, or store it or not (among other things).
- Much of the cholesterol we eat is in the form of CE. It is not absorbed and is excreted by our gut (i.e., leaves our body in stool). The reason this occurs is that CE not only has to be de-esterified, but it competes for absorption with the vastly larger amounts of UC supplied by the biliary route.
- Re-absorption of the cholesterol we synthesize in our body (i.e., endogenous produced cholesterol) is the dominant source of the cholesterol in our body. That is, most of the cholesterol in our body was made by our body.
- The process of regulating cholesterol is very complex and multifaceted with multiple layers of control. I’ve only touched on the absorption side, but the synthesis side is also complex and highly regulated. You will discover that synthesis and absorption are very interrelated.
- Eating cholesterol has very little impact on the cholesterol levels in your body. This is a fact, not my opinion. Anyone who tells you different is, at best, ignorant of this topic. At worst, they are a deliberate charlatan. Years ago the Canadian Guidelines removed the limitation of dietary cholesterol. The rest of the world, especially the United States, needs to catch up. To see an important reference on this topic, please look here.
- Cholesterol and triglycerides are not soluble in plasma (i.e., they can’t dissolve in water) and are therefore said to be hydrophobic.
- To be carried anywhere in our body, say from your liver to your coronary artery, they need to be carried by a special protein-wrapped transport vessel called a lipoprotein.
- As these “ships” called lipoproteins leave the liver they undergo a process of maturation where they shed much of their triglyceride “cargo” in the form of free fatty acid, and doing so makes them smaller and richer in cholesterol.
- Special proteins, apoproteins, play an important role in moving lipoproteins around the body and facilitating their interactions with other cells. The most important of these are the apoB class, residing on VLDL, IDL, and LDL particles, and the apoA-I class, residing for the most part on the HDL particles.
- Cholesterol transport in plasma occurs in both directions, from the liver and small intestine towards the periphery and back to the liver and small intestine (the “gut”).
- The major function of the apoB-containing particles is to traffic energy (triglycerides) to muscles and phospholipids to all cells. Their cholesterol is trafficked back to the liver. The apoA-I containing particles traffic cholesterol to steroidogenic tissues, adipocytes (a storage organ for cholesterol ester) and ultimately back to the liver, gut, or steroidogenic tissue.
- All lipoproteins are part of the human lipid transportation system and work harmoniously together to efficiently traffic lipids. As you are probably starting to appreciate, the trafficking pattern is highly complex and the lipoproteins constantly exchange their core and surface lipids.
- The measurement of cholesterol has undergone a dramatic evolution over the past 70 years with technology at the heart of the advance.
- Currently, most people in the United States (and the world for that matter) undergo a “standard” lipid panel, which only directly measures TC, TG, and HDL-C. LDL-C is measured or most often estimated.
- More advanced cholesterol measuring tests do exist to directly measure LDL-C (though none are standardized), along with the cholesterol content of other lipoproteins (e.g., VLDL, IDL) or lipoprotein subparticles.
- The most frequently used and guideline-recommended test that can count the number of LDL particles is either apolipoprotein B or LDL-P NMR, which is part of the NMR LipoProfile. NMR can also measure the size of LDL and other lipoprotein particles, which is valuable for predicting insulin resistance in drug naïve patients, before changes are noted in glucose or insulin levels.
- The progression from a completely normal artery to a “clogged” or atherosclerotic one follows a very clear path: an apoB containing particle gets past the endothelial layer into the subendothelial space, the particle and its cholesterol content is retained, immune cells arrive, an inflammatory response ensues “fixing” the apoB containing particles in place AND making more space for more of them.
- While inflammation plays a key role in this process, it’s the penetration of the endothelium and retention within the endothelium that drive the process.
- The most common apoB containing lipoprotein in this process is certainly the LDL particle. However, Lp(a) and apoB containing lipoproteins play a role also, especially in the insulin resistant person.
- If you want to stop atherosclerosis, you must lower the LDL particle number. Period.
- At first glance it would seem that patients with smaller LDL particles are at greater risk for atherosclerosis than patients with large LDL particles, all things equal.
- “A particle is a particle is a particle.” If you don’t know the number, you don’t know the risk.
- With respect to laboratory medicine, two markers that have a high correlation with a given outcome are concordant – they equally predict the same outcome. However, when the two tests do not correlate with each other they are said to be discordant.
- LDL-P (or apoB) is the best predictor of adverse cardiac events, which has been documented repeatedly in every major cardiovascular risk study.
- LDL-C is only a good predictor of adverse cardiac events when it is concordant with LDL-P; otherwise it is a poor predictor of risk.
- There is no way of determining which individual patient may have discordant LDL-C and LDL-P without measuring both markers.
- Discordance between LDL-C and LDL-P is even greater in populations with metabolic syndrome, including patients with diabetes. Given the ubiquity of these conditions in the U.S. population, and the special risk such patients carry for cardiovascular disease, it is difficult to justify use of LDL-C, HDL-C, and TG alone for risk stratification in all but the most select patients.
- To address this question, however, one must look at changes in cardiovascular events or direct markers of atherosclerosis (e.g., IMT) while holding LDL-P constant and then again holding LDL size constant. Only when you do this can you see that the relationship between size and event vanishes. The only thing that matters is the number of LDL particles – large, small, or mixed.
- HDL-C and HDL-P are not measuring the same thing, just as LDL-C and LDL-P are not.
- Secondary to the total HDL-P, all things equal it seems smaller HDL particles are more protective than large ones.
- As HDL-C levels rise, most often it is driven by a disproportionate rise in HDL size, not HDL-P.
- In the trials which were designed to prove that a drug that raised HDL-C would provide a reduction in cardiovascular events, no benefit occurred: estrogen studies (HERS, WHI), fibrate studies (FIELD, ACCORD), niacin studies, and CETP inhibition studies (dalcetrapib and torcetrapib). But, this says nothing of what happens when you raise HDL-P.
- Don’t believe the hype: HDL is important, and more HDL particles are better than few. But, raising HDL-C with a drug isn’t going to fix the problem. Making this even more complex is that HDL functionality is likely as important, or even more important, than HDL-P, but no such tests exist to “measure” this.
Did you say “delay?”
That’s right. The question posed above did not ask how one could “prevent” or eliminate the risk cardiovascular disease, it asked how one could “delay” it. There is a difference. To appreciate this distinction, it’s worth reading this recent publication by Allan Sniderman and colleagues. Allan sent me a copy of this paper ahead of publication a few months ago in response to a question I had posed to him over lunch one day. I asked,
“Allan, who has a greater 5-year risk for cardiovascular disease, a 25 year-old with a LDL-P/apoB in the 99th percentile or a 75-year-old with a LDL-P/apoB in the 5th percentile?”
The paper Allan wrote is noteworthy for at least 2 reasons:
- It’s an excellent reminder that age is a paramount risk factor for cardiovascular disease.
- It provides a much better (causal) model for atherosclerosis than the typical age-driven models, and explains why age is an important risk factor.
What do I mean by this? Most risk calculators (e.g., Framingham) take their inputs (e.g., age, gender, LDL-C, HDL-C, smoking, diabetes, blood pressure) and calculate a 10-year risk score. If you’ve ever played with these models you’ll quickly see that age drives risk more than any other input. But why? Is there something inherently “risky” about being older?
Sniderman and many others would argue (and I agree) that the reason age is a strong predictor of risk has to do with exposure to apoB particles — LDL, Lp(a), and apoB-carrying remnants. Maybe it’s because I’m a math geek, but such models just seem intuitive to me because I think of most things in life in terms of calculus, especially integrals, the “area under a curve.”
[I once tried to explain to a girlfriend who thought I wasn’t spending enough time with her that my interest in her should be thought of in terms of the area under the curve, rather than any single point in time. That is, think in terms of the integral function, not the point-in-time function. Needless to say, she broke up with me on the spot (in the middle of a parking lot!), despite me drawing a very cool picture illustrating the difference, which I’ve re-created, below.]
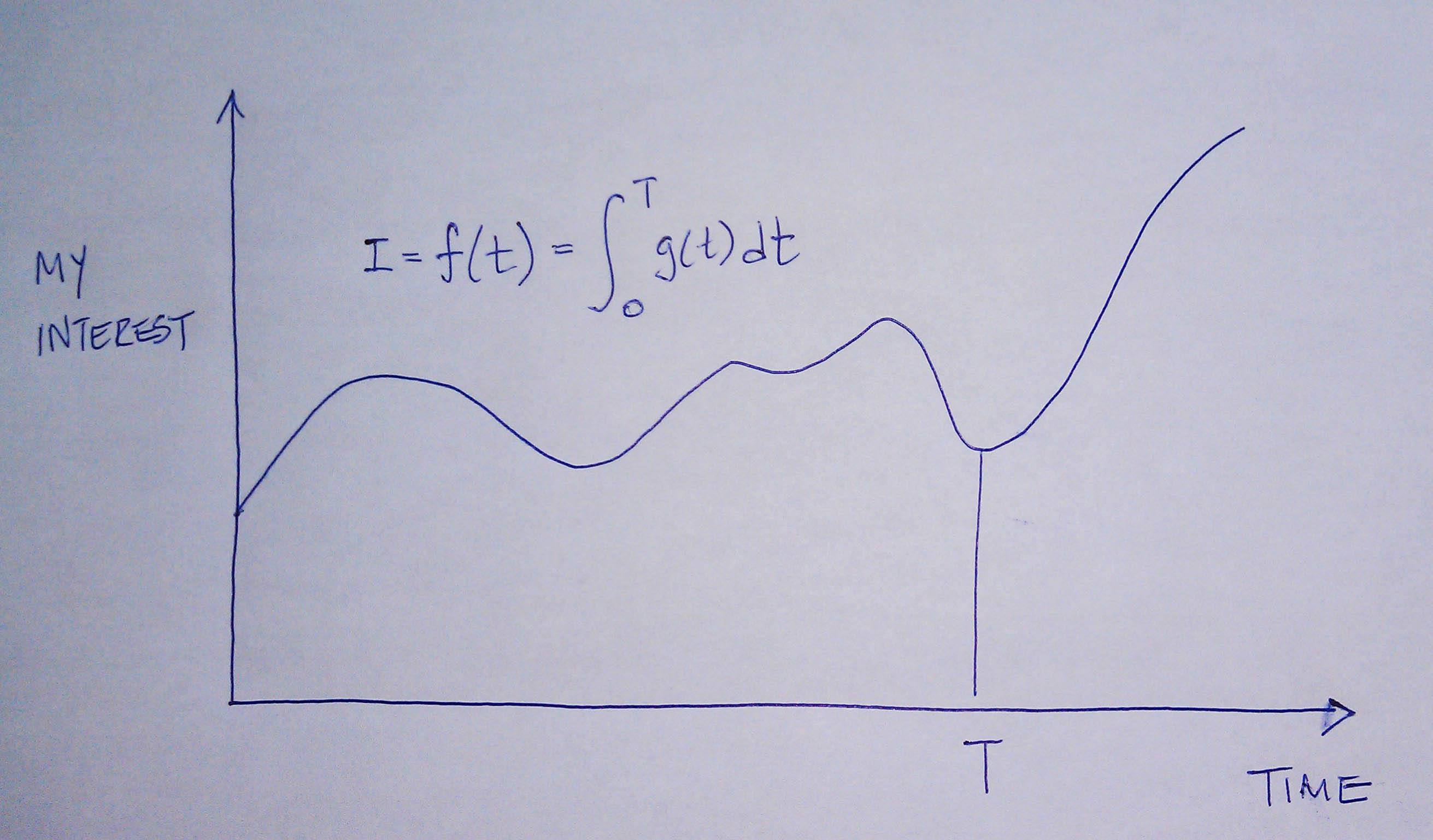
The reason age is such a big driver of risk is that the longer your artery walls are exposed to the insult of apoB particles, the more likely they are to be damaged, for all the reasons we covered in Part IV of this series. [This paper also reviews the clinical situation of PCSK9 mutations which builds a very compelling case for the causal model of apoB particles in the development of atherosclerosis].
What does eating have to do with cardiovascular risk?
So now that everyone is on the edge of their seat in anticipation of this punch-line, let me provide two important caveats.
First, there are no long-term studies – either in primary or secondary prevention – examining the exact question we all want to know the answer to with respect to the role of dietary intervention on cardiovascular disease. There are short-term studies, some of which I will highlight, which look at proxies for cardiovascular disease, but all of the long-term studies (looking at secondary prevention), are either drug studies or multiple intervention studies (e.g., cholesterol-lowering drug(s) + blood pressure reducing drug(s) + dietary intervention + exercise + …).
In other words, the “dream” study has not been done and won’t be done for a long time. The “dream” study would follow 2 randomized groups for many years and only make one change between the groups. Group 1 would consume a standard American diet and group 2 would consume a very-low carbohydrate diet. Furthermore, compliance within each group would be excellent (many ways to ensure this, but none of them are inexpensive – part of why this has not been done) and the study would be powered to detect “hard outcomes” (e.g., death), instead of just “soft outcomes” (e.g., changes in apoB, LDL-C, LDL-P, TG).
Second, everything we have learned to date on the risk relationship between cardiovascular disease and risk markers is predicated on the assumption that a risk maker of level X in a person on diet A is the same as it would be for a person on diet B.
Since virtually all of the thousands of subjects who have made up the dozens of studies that form the basis for our understanding on this topic were consuming some variant of the “standard American diet” (i.e., high-carb), it is quite possible that what we know about risk stratification is that this population is not entirely fit for extrapolation to a population on a radically different diet (e.g., a very-low carbohydrate diet or a ketogenic diet). Many of you have asked about this, and my comments have always been the same. It is entirely plausible that an elevated level of LDL-P or apoB in someone consuming a high-carb diet portends a greater risk than someone on a ketogenic or low-carb diet. There are many reasons why this might be the case, and there are many folks who have made compelling arguments for this hypothesis.
But we can’t forget the words of Thomas Henry Huxley, who said, “The great tragedy of science is the slaying of a beautiful hypothesis by an ugly fact.” Science is full of beautiful hypothesis slayed by ugly facts. Only time will tell if this hypothesis ends up in that same graveyard, or changes the way we think about lipoproteins and atherosclerosis.
The role of sugar in cardiovascular disease
Let’s start with what we know, then fill in the connections, with the goal of creating an eating strategy for those most interested in delaying the onset of cardiovascular disease.
There are several short-term studies that have carefully examined the impact of sugar, specifically, on cardiovascular risk markers. Let’s examine one of them closely. In 2011 Peter Havel and colleagues published a study titled Consumption of fructose and HFCS increases postprandial triglycerides, LDL-C, and apoB in young men and women. If you don’t have access to this journal, you can read the study here in pre-publication form. This was a randomized trial with 3 parallel arms (no cross-over). The 3 groups consumed an isocaloric diet (to individual baseline characteristics) consisting of 55% carbohydrate, 15% protein, and 30% fat. The difference between the 3 groups was in the form of their carbohydrates.
Group 1: received 25% of their total energy in the form of glucose
Group 2: received 25% of their total energy in the form of fructose
Group 3: received 25% of their total energy in the form of high fructose corn syrup (55% fructose, 45% glucose)
The intervention was relatively short, consisting of both an inpatient and outpatient period, and is described in the methodology section.
Keep in mind, 25% of total energy in the form of sugar is not as extreme as you might think. For a person consuming 2,400 kcal/day this amounts to about 120 pounds/year of sugar, which is slightly below the average consumption of annual sugar in the United States. In that sense, the subjects in Group 3 can be viewed as the “control” for the U.S. population, and Group 1 can be viewed as an intervention group for what happens when you do nothing more in your diet than remove sugar, which was the first dietary intervention I made in 2009.
Despite the short duration of this study and the relatively small number of subjects (16 per group), the differences brought on by the interventions were significant. The figure below shows the changes in serum triglycerides via 3 different ways of measuring them. Figure A shows the difference in 24-hour total levels (i.e., the area under the curve for serial measurements – hey, there’s our integral function again!). Figure B shows late evening (post-prandial) differences. Figure C shows the overall change in fasting triglyceride level from baseline (where sugar intake was limited for 2 weeks and carbohydrate consumption consisted only of complex carbohydrates).
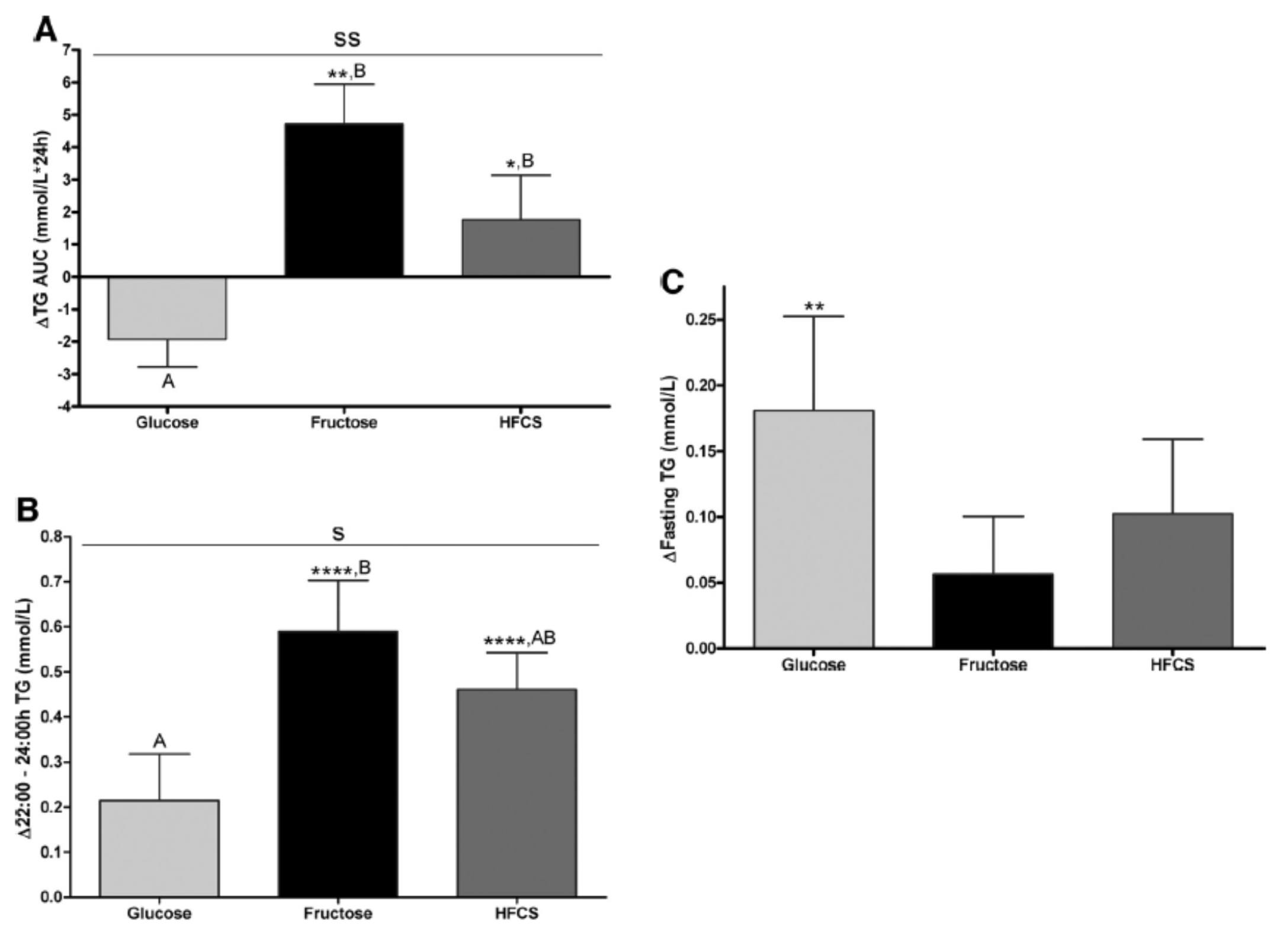
The differences were striking. The group that had all fructose and HFCS removed from their diet, despite still ingesting 55% of their total intake in the form of non-sugar carbohydrates, experienced a decline in total TG (Figure A, which represents the daily integral of plasma TG levels, or AUC). However, that same group experienced the greatest increase in fasting TG levels (Figure C). Post-prandial TG levels were elevated in all groups, but significantly higher in the fructose and HFCS groups (Figure B). The question this begs, of course, is which of these measurements is most predictive of risk?
Historically, fasting levels of TG are used as the basis of risk profiling (Figure C), and according to this metric glucose consumption appears even worse than fructose or HFCS. However, recent evidence suggests that post-prandial levels of TG (Figure B) are a more accurate way to assess atherosclerotic risk, as seen here, here, and here. One question I have is why did the AUC calculations in Figure A show a reduction in plasma TG level for the glucose group?
The figure below summarizes the differences in LDL-C, non-HDL-C, apoB, and apoB/apoA-I.
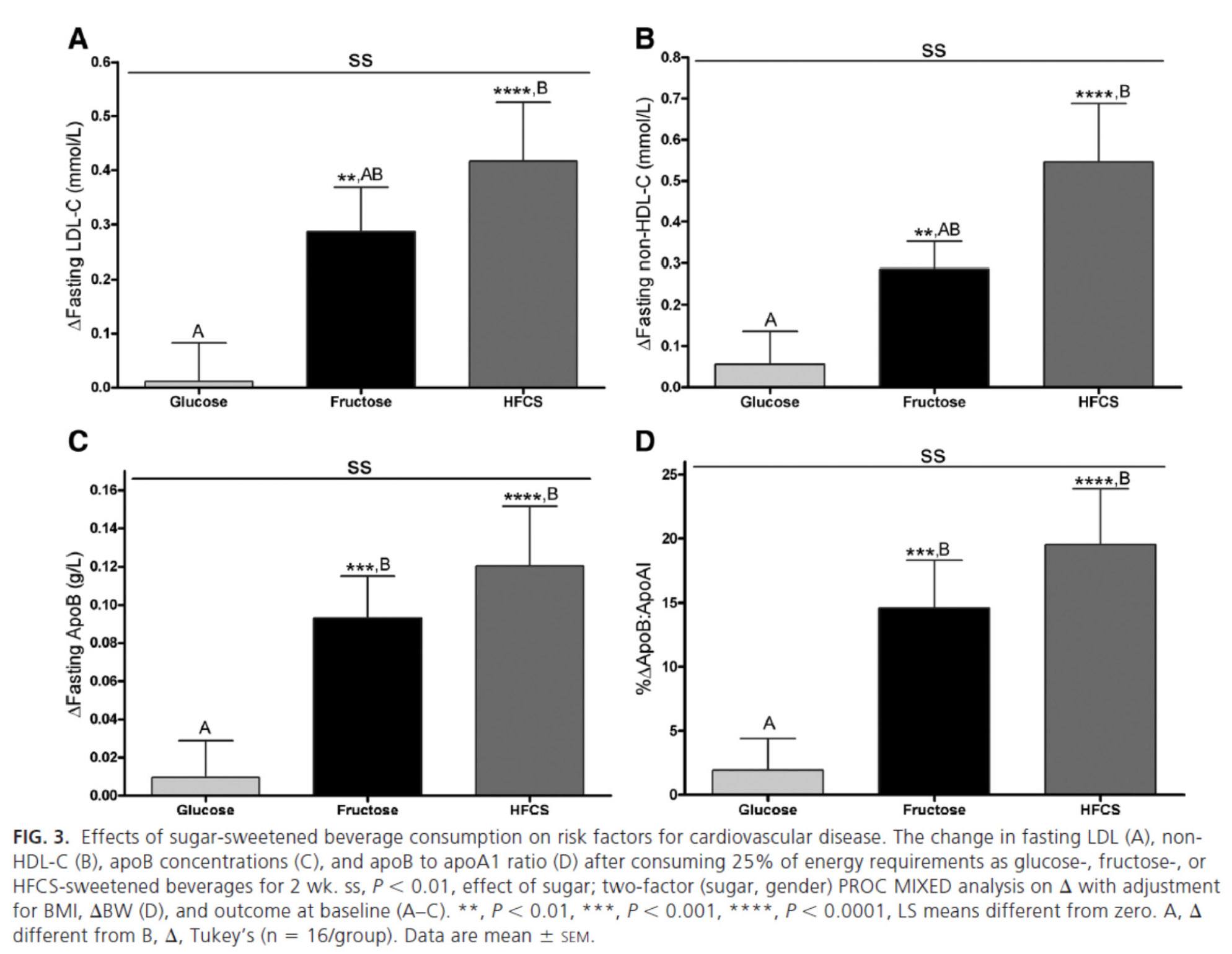
Again, the results were unmistakable with respect to the impact of fructose and HFCS on lipoproteins, and by extension, the relative lack of harm brought on by glucose in isolation. [Of course, removal of glucose and fructose/HFCS would have been a very interesting control group.]
One of the simultaneous strengths and weaknesses of this study was the heterogeneity of its subjects, who ranged in BMI from 18 to 35, in age from18 to 40, and in gender. While this provided at least one interesting example of age-related differences in carbohydrate metabolism (older subjects had a greater increase in triglycerides in response to glucose than younger subjects), it may have actually diluted the results. There were also significant differences between genders in the glucose group.
What was most interesting about this study was the clear difference between the 3 groups that was not solely a function of fructose load. In other words, the best outcome from a disease risk standpoint was in the glucose group, while the worst outcome was not in the all-fructose group, but in the 50/50 (technically 55/45) mixed group. This is a very powerful indication that while glucose and fructose alone can be deleterious in excess, their combination seems synergistically bad.
The role of saturated fat in cardiovascular disease
In the next week or two I’ll be posting an hour-long comprehensive lecture I gave at UCSD a few weeks ago on this exact topic. Rather than repeat any of it here, I’ll highlight one study that I did not include in that lecture. The study, Effect of a high saturated fat and no-starch diet on serum lipid subfractions in patients with documented atherosclerotic cardiovascular disease, published in 2003, treated 23 obese patients (average BMI 39) with known cardiovascular disease (status post coronary artery bypass surgery and/or stent placement) with a high-fat ketogenic diet. Because the study was free-living and relied on self-reporting, not all subjects had documented levels of elevated serum B-OHB. However, the subjects were instructed to avoid starch and consume 50% of their caloric intake via saturated fat, primarily in the form of red meat and cheese. There were no restrictions on fruits and vegetables, which may have accounted for the observation that not all subjects were ketotic during the 6-week intervention. In total, only 5 of the 23 patients achieved documented ketosis.
All of the subjects were on statins and entered the study at a goal LDL-C level target of 100 mg/dL, which may have been the only way the authors could get the IRB to approve such a study.
The table below shows the changes in lipoprotein fractions following the intervention (there was no control group):
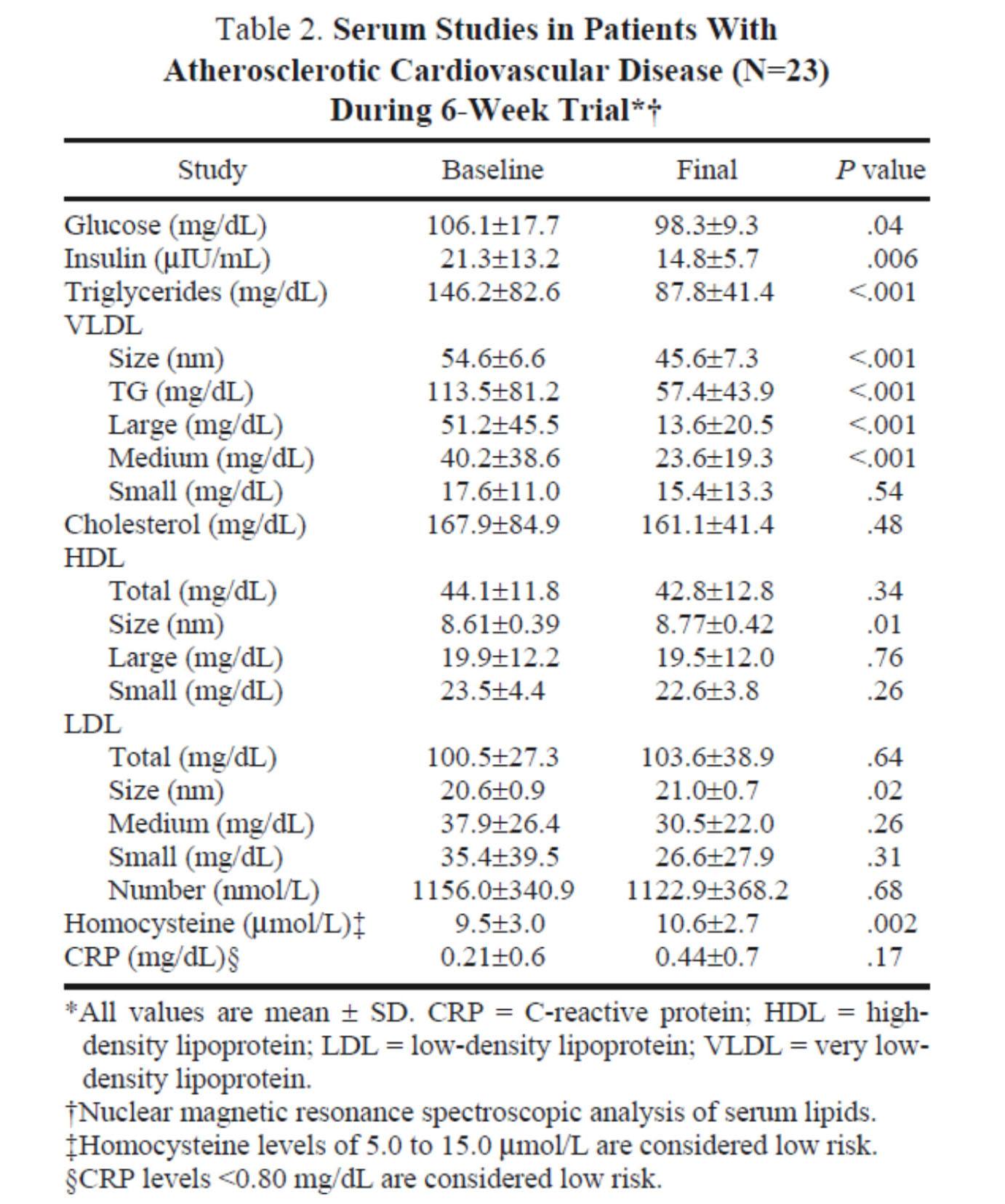
This study was conducted during the height of the “outcry” over the Atkins diet. While most doctors reluctantly agreed that Dr. Atkins’ diet could reduce body fat, most believed it was still very dangerous. In the words of Dean Ornish, “Sure you can lose weight on a low-carb diet, but you can also lose weight on heroin and no one would recommend that!”
Fair point. In fact, the authors of this study acknowledged that they “strongly expected” this dietary intervention to increase risk for cardiovascular disease, which is why they only included subjects on statins with low LDL-C. However, as you can see from the table above, the authors were startled by the results. The subjects experienced a significant reduction in plasma triglycerides and VLDL triglycerides, without an increase in LDL-C or LDL-P. In fact, LDL size and HDL size increased and VLDL size decreased – all signs of improved insulin resistance. Furthermore, fasting glucose and insulin levels also decreased significantly. The mean HOMA-IR was reduced from 5.6 to 3.6 (normal is 1.0) and TG/HDL-C from 3.3 to 2.0 (normal is considered below 3, but “ideal” is probably below 1.0) in just 6 weeks. Taken together, these changes, combined with the dramatic change in VLDL size, suggest insulin resistance was dramatically improved while consuming a diet of 50% saturated fat!
As all of these patients were taking statins, we’re really robbed of seeing the impact of this diet on LDL-P, which did not change. Also, CRP levels rose (though not clinically or statistically significantly).
Putting it all together
It is very difficult to make the case that when carbohydrates in general, and sugars in particular, are removed or greatly reduced in the diet, insulin resistance is not improved, even in the presence of high amounts of saturated fats. When insulin resistance improves (i.e., as we become more insulin sensitive), we are less likely to have the signs and symptoms of metabolic syndrome. As we meet fewer criteria of metabolic syndrome, our risk of not only heart disease, but also stroke, cancer, diabetes, and Alzheimer’s disease goes down.
Furthermore, as this study on the Framingham cohort showed us, the more criteria you have along the spectrum of metabolic syndrome, the more difficult it becomes to predict your risk, due to a widening gap in discordant risk markers, as shown in this figure.
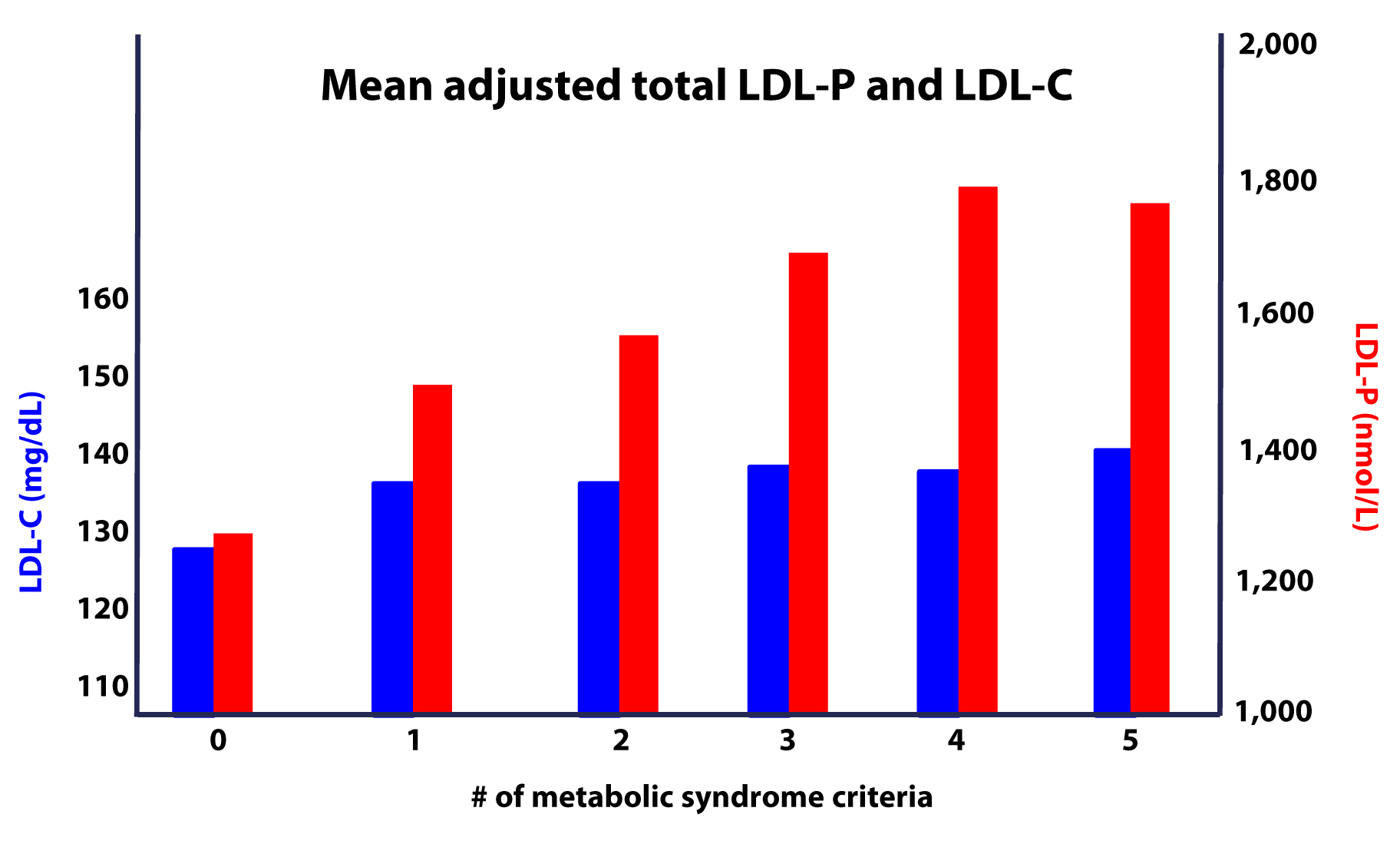
As I noted at the outset, the “dream” trial has not yet been done, though we (NuSI) plan to change that. Until then each of us has to make a decision several times every day about what we will and won’t put in our mouths. Much of this blog is dedicated to underscoring the impact of carbohydrate reduction on insulin resistance and metabolic syndrome.
The results of the trials to date, combined with a nuanced understanding of the lipoprotein physiology and their role on the atherosclerotic disease process, bring us to the following conclusions:
- The consumption of sugar (sucrose, high fructose corn syrup) increases plasma levels of triglycerides, VLDL and apoB, and reduces plasma levels of HDL-C and apoA-I.
- The removal of sugar reverses each of these.
- The consumption of fructose alone, though likely in dose-dependent fashion, has a similar, though perhaps less harmful, impact as that of fructose and glucose combined (i.e., sugar).
- The addition of fat, in the absence of sugar and starch, does not raise serum triglycerides or other biomarkers of cardiovascular disease.
- The higher the level of serum triglycerides, the greater the likelihood of discordance between LDL-C and LDL-P (and apoB).
- The greater the number (from 0 to 5) of inclusion criteria for metabolic syndrome, the greater the likelihood of discordance between LDL-C and LDL-P (and apoB).
I would like to address one additional topic in this series before wrapping it up – the role of pharmacologic intervention in the treatment and prevention of atherosclerotic disease, so please hold off on questions pertaining to this topic for now.

Hi Peter-
Read this post awhile back, it has never quite sat right. Your 25 year old vs.75 year old risk example seems to imply that the area under the curve is unchangeable. That is, the damage done to arterial walls via particles is purely cumulative, and cannot be reversed or healed? What about a 65 year old with great numbers? 55 year old? 45? Perhaps I am reading too much into a high level population model on this one example? It is a thought provoking question-…
It’s probably not true in absolute, but on average. Of course, drugs and (the right) lifestyle changes will improve endothelial injury.
Dear Dr. Attia,
thanks a lot for letting laypeople like me participate of both your personal journey and a lot of medical facts/news completed with explanations, so that also not-medically-trained people are able to follow.
I really do appreciate your enthusiasm and your wisdom as well as your generousity in sharing your insights with the world.
I´m an Austrian teacher and I´ve read nearly all your posts within the last weeks.
I started LCHF just a while ago and I´ve got a question, which hasn´t be posted yet (as far as I can see).
I wondered, if it is thinkable that a nutrition being high in fat leading to plaques in the artery-walls WITHOUT
uppening the blood-lipids (aop-b-particle- number/LDL-C/total cholesterol)?
What I´m wondering exactly is: Is it possible, that all blood-lipids- expecially the number of apo-b- particles, but also the LDL-C or even the triglycerides – are perfectly fine : No signs of the usually risk-factors for a cardio-vascular-desease. And that despite this- at the same time plaques are buildung within the artery-walls, even though the blood-lipids are perfect?
Might it be possible, that the plaque-building goes on unwarned by blood-lipids?
Or that there might be different risk-markers with high-fat nutrition than with a low-fat-nutrition?
Yours Eva
I guess anything is possible, but it would be less likely, especially if specific inflammatory markers were also normal (e.g., LpPLA2, MPO).
“7. Eating cholesterol has very little impact on the cholesterol levels in your body. This is a fact, not my opinion. Anyone who tells you different is, at best, ignorant of this topic. At worst, they are a deliberate charlatan.”
This is rubbish! I went on Dr. Esselstyn’s diet almost three years ago and it cut my LDL-C in half. My Cardiologist had me on a statin, but after awhile I stopped taking it completely for two months and then rechecked–still was half what it was before. I think you folks are the charlatans! I restarted on a different statin with less side effects and at a very low self-imposed dose (5 mg pravastatin/day)–just to hedge my bets. Checked it again last week and it’s 67. When I had my stent it was 134. Folks, go to http://www.heartattackproof.com and turn your lives around!
Your statement is no way a refutation of mine. Read my statement carefully. I did not say what you eat does not impact the cholesterol levels in your body. I said eating cholesterol… big difference, my friend.
Hi Dave,
In my case a low-carb diet is enough to have perfect blood lipids. You seem to be eating high-carb and taking statins to try to fix the problems carbs create. I am sorry for you and I hope some day you can get rid of those drugs.
I would recommend reading “Systematic review and meta-analysis of clinical trials of the effects of low carbohydrate diets on cardiovascular risk factors ” if you want to know what science has to say about improving your health without drugs.
Have a nice day
C’mon, doc. You know people equate eating cholesterol with eating animal products. You’re playing word games here.
And Vicente, that study does nothing to move me. I agree that sugar and processed grains like white flour are no good, so if you cut that out of an animal product diet you’ll naturally fare better. No need to feel sorry for me, my endothelium’s in a lot better shape than the SAD diet I ate most of my life left it in. That’s what’s important, not eating to achieve good “numbers” (of which mine are substantially better just the same). If you don’t eat stuff that injures your arterial lining, your numbers mean little. What good is a high HDL if it really just means your liver is working overtime to mop up the residue from your bad eating habits?
I’m outta here…
Hi Dave,
with a low-carb diet I have better blood lipids (raised HDL, same LDL, lower triglycerides) and normal transaminases. My TG/HDL is 0.7
My numbers are amazing, but you say this is in fact bad for me… You mean that now that my transaminases are ok, my liver and kidneys are indeed suffering? My high HDL “really just means my liver is working overtime”? While my health parameters improve my health deteriorates? Are you serious?
I trust the numbers and the facts: 23 clinial trials using low-carb diets say my diet is healthy. But you “know” my diet is “bad eating”…
Have a nice day
Dave: After a year and a half or so of following Peter Attia’s blog, I can say with pretty good confidence that one thing he does NOT do is play word games. That’s why I count on him as a trustworthy source of information. Which is not to say that you or I or anyone should accept what he presents uncritically (in fact, Attia would caution against doing so), but it’s possible to disagree without making ad hominem attacks on his integrity.
I agree Kelley, I have been foolowing Peter’s blog for three years and it has been an incredible journey.
Peter, I have finally gotten to the consciously unconscious stage and would like to start a light weight lifting routine for women over 50. I don’t know where to turn though. Any suggestions. I currently use the “Lower Back Basics” and ” Flexibility Training” by Brian Doftman. Thanks in adavance for any suggestions. Have a Great Father’s Day!
Thank you, Ellen. I’d recommend the book by Doug McGuff (Body By Science) and then a well-certified super slow coach to guide you.
Hi Peter, I love what your are doing and the pasion you do this with is unique.
I meant passion not “pasion”. sorry.
I would like to get your comment on my lipid profile. You mentioned in your talks that you do not know whether a low carb diet that increases LDL is a worry when all other metabolic risks are low. I am one of those, normal blood pressure (115/75), low triglycerides (0.9) High HDL 2.2, and very high LDL: 6 That is 230 for you in the USA), almost all is large ‘non-atherogenic’ (I’m in New Zealand) However the shock is a very high oxidised LDL 2500.0 + ng/ml (reference range: 90 – 133.2)- a test not normally done in USA, I got mine done through a lab in Germany.
I emailed Dr Dayspring, who said that it is likely the saturated fat in my diet “Your case is representative of many who pursue the high fat, low carb dietary methods. Due to individual genetic, some use the saturated fat as a substrate to significantly increase cholesterol synthesis – measuring the penultimate sterols in the cholesterol synthesis pathway ( desmosterol or lathosterol) in serum would confirm this.”
As you said we just dont know if this is a problem or not. My husband also had his tested, and depite a similar diet, his LDL is also high (5), but virtually no oxLDL. Results from the lab (imupro) are in PDF format. I’d be happy to forward them to you if you are interested.
As you can imagine – I’ve changed my diet – taken out coconut cream and oil which has caused problems for others LDL, and increased root veg and berry carbs. and added some supplements. I’m keeping track of everything so I can see what effect diet has.
Regards
Julianne
Hi Peter,
Enjoy your posts, great stuff! Wanted to get your thoughts on the recent news about adverse effects of Niacin on hearth health. From your understanding does it really decrease LDL-P and increase HDL?
https://www.foxnews.com/health/2014/07/17/dont-take-niacin-for-heart-health-docs-warn/
Thanks
Mike
Here’s the email I sent Tom Dayspring a week ago when I read the NEJM study:
“Tom, So I read the NEJM piece (but have not seen your commentary yet, so forgive me if I’m repeating what you have written there)…
This struck me as a study that asked the wrong question!
They take a group of patients with already very low LDL-C (and presumably, though not a given, low LDL-P) and add one more agent to try to eke out a bit more LDL-C (and presumably LDL-P) reduction.
But the agent they chose—-niacin-—is known to worsen IR.
They do this in a patient population that is either all T2D or pre-diabetic. At best, they are “just” IR.
So the trade-off is lousy. The upside-—LDL-P reduction—-is almost non-existent, yet the downside-—worsening IR-—is huge.
Maybe I’m being too critical of the study. If this use of Niaspan is “common,” then it’s a good thing to have studied it the way they did, but it seems odd.”
LDL-P
!. If 100 hundred people enter the hospital or die having a heart attack (soft plauqe eroption) – how many of these people will have high LDL-P – ?
a. A cofounder to this might be intermittent high LDL-P or high levels at some prior period – but aside from this possibility –
they all should have high LDL-P –
The number better be at least 50 – less than this and your LDL-P theory becomes not only useless and outright misleading – unless these is the best predictive available –
I suppose or a least hope – the number is between 50 and a 100 hundred –
Show me some numbers
I have a feeling this may not be pretty –
How can this be done, Jeff, when about 50% of the time some presents with CAD, the first sign is sudden death? Furthermore, with so few people receiving advanced testing, the LDL-P info would be inconsistent.
Look at the LDL-C data in one of these post (can’t recall which one) to see that distribution–LDL-C is not predictive in the hospital admitted patient population.
If you look at studies that do use LDL-P testing, including this one (Atherosclerosis 235 (2014)) that came out a few weeks ago, mortality always tracks higher with LDL-P than any other metric.
Hi Jeff,
1) let’s assume 5% of the population has a high LDL-P.
2) let’s assume 25% of the people that die having a heart attack has a high LDL-P.
Wouldn’t you say a high LDL-P is a risk factor?
Hi Peter,
Thanks for the incredible posts and video lectures. Few questions, please:
How does one lower ApoB? I’ve read that Tangeretin and Nobiletin inhibits ApoB production, but not sure that is addressing the cause. I’ve been Paleo for 2 years and my measured ApoB is 126 and apparently under 109 is acceptable range. ApoAI is 129, so the ratio is 0.98 where acceptable is <0.92. My Lp(a) is 4. So I'm struggling to understand why the ApoB is out of range.
Also, how would one reverse endothelialc damage. If ApoB is lowered, can atherosclerosis be reversed? I supplement with theracumin to support this.
Lastly, please could you let us have your viewpoint on Lp-pla2 – does this test have any merit? My score is 194 under the reference range of 200 which is a relief after my ApoB level.
Sincere thanks,
Andy
HI Peter – sorry for the bump, but your viewpoint would really be appreciated. Cheers!
HI Andy – did you find any response to this. I have similar questions.
Thanks, Quentin
Hi Peter,
Let me first say…..I’m a huge fan!
My question for you is:
If a person has metabolic syndrome, has had elevated triglycerides for some time (thus increased lipoproteins, boats), when this person adopts a nutritional ketotic diet and trigs fall through the floor does the measurement of HDL, LDL and overall cholesterol become elevated?…number of boats still the same but now carrying cholesterol (the intended cargo). If so, how long before the extra boats are decommissioned?
Cheers,
Rob
There may be a transient effect, but typically, a reduction in TG is matched by a rise in HDL-C within about a month or two. LDL-C is more variable, can go up or down depending on fat content of diet. VLDL-C also goes down typically.
Hi Peter,
I am very confused do to the changes in my lipid profil in the last couple of months. I am 30 years old, I train on a daily basis ( I’m a kettlebell trainer, and I do crossfit and run 5k 2 a week). Everyday I try to be active. I weight 54 kg and my height is 1.60 cm and fat percentage is about 20%.
Now, I’ve started Paleo diet on January this year and did my blood check at the same time and it turned out great (hdl 113, LDL 80). My paleo was less then 100 gr carbs per day. i decided to try ketogenic diet do to a lot of reading and quriuos on the way it will effect my training and body. On April I went into ketosis (under 20 gr carbs per day). I checked every day with blood strips my ketone and glucose. After 2 month I did blood checks again and I was very confused – my hdl rose to 154, but my LDL rose to 270. Glucose was 69, no CRP, thyroid ok, and triglycerid went just a little up. After a month I did blood checks again and my LDL rose again to 300. I checked my apo b and it was high (188), vldl was normal.
I’ve decided to get out of ketosis and back to paleo ( I eat 80-100 gr fat, 70-100 gr carbs only from non starch vegetables and 1.2-1.5 per kg protein per day).
I can’t understand the dramatic changes in my blood profil. I will state that before paleo I ate low fat low carb – a lot of vegetables, low fat dairy, no red meat, legumes, eggs , fish and chicken.
I wonder if ketosis act different on active people. I’m quiet stressed at the moment, I’m used to be very healthy (as the modern age see it), and my doctor went nuts from my latest blood check. I really appreciate hearing your view on this.
Much appreciate
Rinat-ya
this may help you
https://perfecthealthdiet.com/2011/03/answer-day-what-causes-high-ldl-on-low-carb-paleo/
Hello Dr. Attia,
Can you suggest a source for arachidonic acid for someone who cannot consume animal products due to health issues such as testing positive for the ApoE 3/4 genotype test?
Thank you so much.
Jim
Peter – Do you know of any more in-depth discussion of the application of Sniderman’s “Causal Exposure Model”? I am unclear how to apply a rating to something eg “intensity of smoking”. I also would love to get some context on interpreting the model’s resulting number. Great article.
Thank you for all your hard work, it is Much appreciated.
Sniderman has written a lot about this topic. Check his other work.
would like to address one additional topic in this series before wrapping it up – the role of pharmacologic intervention in the treatment and prevention of atherosclerotic disease, so please hold off on questions pertaining to this topic for now.
Where is this post?
Still in my brain…
Hi Peter,
wanted to ask which test is a better predictor of CVD: LDL-P/Apo B or LP-PLA2 ?
Thanks
The former.
Hello Peter,
I started my nutritional research / study / and dietary journey almost 6 months ago. I can’t even put words to convey how impressed I am with both you as a human being and the quality of work you put forth!!!
I just finished this series and have a technical question that I’m hoping you can answer. I fully understand how CHO consumption affects TG levels in the bloodstream. ie… increased presence of insulin which promotes the process of 3 free fatty acids combining to a glycerol molecule to form TG… combined with the insulin also preventing/blocking the enzyme necessary to break the TG back down from doing it’s job.
What I haven’t figured out is this… what is it about the presence of insulin or how is it altering the lipid profiles? ie… higher LDL-P
In reading your work, stuff from Gary and also Tim Nokes I readily accept and fully believe that a LCHF and/or fully ketogenic diet definitely alter lipid profiles for the better but I just haven’t been able to figure out why from a technical standpoint … the way I understand it’s effects on TG levels.
If I had to guess, I would guess it has something to do with the lipid transport system, the shedding and/or exchanging of TGs and cholesterol during this dynamic process, and the higher levels TG present due to the constant presence of insulin somehow influencing the lipid profiles but I am in no way sure about that! Am I on the right track or is it something entirely different?
Thanks!!!
Vance
Biggest change may be reduction of TG, which increases CE-carrying capacity of lipoproteins–more room for carge, fewer boats.
Peter, Mind if I post a PDF containing all 9 posts. It has all links turned into endnotes so people can print the entire series (or read on an e-reader) easily?
Sure.
Here it is: https://drive.google.com/file/d/0B5klE_D9KbB-Q1Fubkg1R092U28/view?usp=sharing
Nicely done James! I’ve been hacking my own consolidated version for a while now.
WIth an NMR LDL-P over 3200, I am profoundly interested in Peter’s work.
Peter – Your work is great in this field. Lots of other individuals are all about selling their supplements, books, etc. At some point you need to sell something to get some return for your time other than admiration and respect. Maybe that is enough.
I’ve listened to your video on Cholesterol several times and now I fully understand it. Its great. Your question at the end, I’ve ignored for a long time and didn’t think it applied to me. Now I think it does apply to me. I’ve been on a keto diet for 5 months. I’m an athlete like yourself and continued to exercise on the keto diet as I had in the past. I felt like I could recover faster but I would feel sluggish at times. Dr. Phinney recommended to me to take the salts and potassium prior to workouts. For example I would swim for 50 minutes in the morning and jog 5 miles or lift weights at lunch time. Do that 4 times a week followed with a long bike ride on Sundays (4 hours – not eat anything on the ride – bonk proof, amazing). I’m 6’2″ 177lbs. I tracked my foods ~2500 cals per day, 120g protein, under 50g total carbs a day (more like 30 total carbs). I’ve improved my body composition. Then I got my blood panel tested. I’m 41 years old. As a carb eater TC 206, LDL 125, HDL 58, Tri 117. Keto diet TC 327, LDL 211, HDL 101, Tri 74. I’ve had it measured 3 times now and they are consistent. My Tri are down to 46. Amazing. My primary care physician and a Stanford cardiologist have been going nuts about my levels. I’ve recently had blood drawn for a LDL-p count and a thyroid examine. I’m waiting for the results. My fasting blood glucose is 105 or 107. It used to be in the high 80s or 90s. I measured my ketones via blood and they were at .5-.7. Never really higher. However after a 4 hour ride they would be at 1.7 or 1.9 and my blood glucose would be at 70. Why is my LDL so high? Any ideas? Why my Blood Glucose so high? A nutritionist at Stanford insists that I up my carbs to 100g or 150g.
Addressed in one of the posts I wrote about the interplay of exercise and ketosis.
If I could just butt in… Volek and Phinney’s book has a chapter on TC and measurements. Short answer: very possible your LDL and TC aren’t measured correctly. If your triglycerides are 46 at this point and your doctors are using the Freidewald equation, your situation may be similar to an example Ph. and V. give, of a man with triglycerides of 55 and LDL-C of 172 through Friedewald, which turned out to be 126 upon two separate direct methods of measurement. So, yeah, bear that in mind. Also, LDL-P, as far as I gather, is by far the more important yardstick.
@Chris Stout: I experienced a very similar change to your changes in cholestrol numbers when I switch to NK this summer. Like you, I exercise regularly and hard. I scoured the 2 postings on exercise and ketosis, but still am puzzled why my glucose and HA1c numbers went UP despite significant increase in cholestrol (total, LDL and HDL) and decrease in Tri. Did you have any luck deciphering this puzzle?
I have read that in 25% of the people the dietary cholesterol adds to the blood cholesterol. Therefore, in some people dietary cholesterol makes a difference. Hence, at least for these people, Odiet or Esselstyn diet may make a difference.
Closer to 10-15% of cholesterol is absorbed.
Awesome post…..my cholesterol is too low so this was perfect.
Still figuring how to raise it. Any other tips?
Also the use of a graph to demonstrate your interest level in a girlfriend was classic. You got off easy my friend. 😉