After last week’s deep dive into the relationship between LDL cholesterol, systolic blood pressure, and cardiovascular disease, I thought it would not hurt to go a bit deeper in the weeds. You’ll recall that atherosclerosis takes the greatest proportion of lives when people are in their later years, but it’s a disease that takes a long time to evolve, starting in the first two decades of life. That’s right, the process begins almost as soon as you’re born. By the time you’re in your 30s, and certainly into your 40s, there is ample evidence of the disease at autopsy.
The etymology of atherosclerosis derives from athero—meaning a gruellike substance, and sclerosis—meaning a hardening. This porridge of oxidized sterols, lipids, cholesterol, macrophages, calcium, fibrin, and other cellular materials make up the lesions, or plaques, within the walls of the arteries. Influenced by multiple risk factors, the atherogenic process begins early in life and slowly progresses until the plaque narrows, erodes, or ruptures causing ischemic clinical events (e.g., fatal or non-fatal heart attack or stroke).
If there is one benefit to a disease that takes so long to progress from microscopic (i.e., subclinically, at the cellular level) to macroscopic (i.e., clinically, at the organ level), it’s this: there is ample time to do something about it. When you think of atherosclerosis, think of saving for retirement. It’s a multivariate compounding problem that takes time. What determines the outcome?
- At what age do you want to retire?
- Will you inherit any money or is there some other windfall awaiting you?
- How much do you plan to spend after you retire? (And how long do you expect to live to spend it?)
- Have you budgeted for catastrophic loss (e.g., 10 years of high-priced assisted living at the end of life)?
- How much do you make before you retire?
- How much can you save during that time?
- What is the net-of-fees, after-tax rate of return on that invested capital?
- At what age do you start saving/investing?
Given that I’m not a financial planner, I’m sure some of you will mock the simplicity of my analogy, but I think you get the point. And you’ll hopefully appreciate that as you manipulate one variable, others need to flex in response. For example, the earlier you want to retire and/or the more you want to spend in retirement and/or the later you wait to start saving, guess what? The more you need to save and/or the higher the rate of return you’ll need. I could go on and on with permutations, but you already know this.
How does this apply to atherosclerosis?
- How old do you want to be before atherosclerosis goes from being subclinical (i.e., no risk of hurting you) to clinically relevant?
- What is your family history of atherosclerosis?
- Do you smoke or have high blood pressure?
- Is your Lp(a) elevated?
- On a scale of 1-10, how metabolically healthy are you?
- What is your current burden of disease?
- What are your biomarkers for atherosclerosis?
- What is your appetite for pharmacologic intervention?
- Do you have contraindications to such interventions?
- How soon are you willing to start such interventions?
I could go on, but I think you get the point. One concern I have with 14-minutes-per-year medicine is that not enough people ask these questions. Especially the first one, believe it or not. Why is this first question so important? No one, not a soul, is immune to this disease, even the genetically gifted centenarians who will outlive us all. So it absolutely matters that you handicap question #1. If you’re interested in longevity—both the lifespan and healthspan part of the equation—it means you want to live longer. That means you want to delay death. That means you want to delay the onset of the most inevitable disease to our species.
Which brings us to the point of today’s post. How can one assess the risk of an individual at a point in time? I’ve written extensively on some of these, but today I want to shine a light on #6—what is the current burden of atherosclerosis?
In fact, I want to shine that light on a subset of the population: those without any clinical evidence of disease (i.e., those who have not already had a heart attack or shown symptoms of heart disease, such as chest pain on exertion). Think of this as a backward-looking proxy for risk (versus biomarkers, which are a forward-looking proxy for risk). If the biomarkers tell you how dangerous your neighborhood is, a backward-looking test will tell you if anyone has tried to break into your home.
There is a tool that is gaining more recognition from academics, guidelines, clinicians, and patients called a coronary artery calcium (CAC) scan. The CAC scan, using computed tomography (better known as CT), assesses the presence and extent of coronary artery calcification (calcium being radio-opaque). The test reports on location and quantification of calcium within the three main coronary arteries and yields a numerical CAC score in a metric called Agatston Units (named after the tool’s developer, Arthur Agatston). A patient with no calcification will have a score of “0” which escalates as vascular calcification increases. A score of “1” and above indicates the presence of calcification, most often in the form of hard plaque. Importantly, and discussed in more detail below, soft plaques can escape detection using a CAC scan. CAC score results fall into four categories which correlate with the severity of disease and range from no disease to severe disease.
Table 1. Categories of CAC scores.

When I talk about CAC, I often use an analogy of atherosclerosis as a crime scene involving breaking, entering, and vandalizing. A criminal went into your house while you were on vacation and did some damage to your home, damage that was somewhat irreparable in that you couldn’t repair your home back to a state where you never would’ve known there was a break-in. Holes in the walls of your home needed to spackle to patch them up. The repair work left clues of damage. In the case of atherosclerosis, a lesion is a damaged artery, and the calcium deposits are a sign of repair to the artery.
A CAC score above zero tells you that there’s been a bad enough break-in to require repair. However, a lot can go on in the disease process leading up to that point that goes unnoticed by a CAC scan. Additionally, a CAC scan does not necessarily identify the plaques that might do the most damage.
A 2013 study, for example, looked at a group of 17 patients who died from an acute myocardial infarction (AMI) and compared them to 15 age-matched controls without a history of cardiovascular disease (CVD). The investigators studied 960 coronary segments in these patients and found calcification in 47% of the segments in the AMI group and in 24.5% in the controls. The calcification was not correlated with the presence of unstable plaques. The study’s conclusions are summed up by the article’s title: “Coronary calcification identifies the vulnerable patient rather than the vulnerable plaque.”
In other words, more than half of the lesions leading to sudden death in the AMI group exhibited little to no calcium. Unstable plaques, the ones more likely to cause grave damage, showed a lower degree of calcification compared to stable plaques. A low to zero CAC score suggests a lower risk of future events, but it does not mean a zero risk of future events. A CAC score is predictive (multiple clinical studies of varied populations have validated the CAC scan as a valuable risk-assessing tool as the extent of CAC accurately predicts 15-year mortality in asymptomatic patients) and diagnostic, certainly a tool worth having in the toolbox, but it doesn’t tell you what else is under the hood.
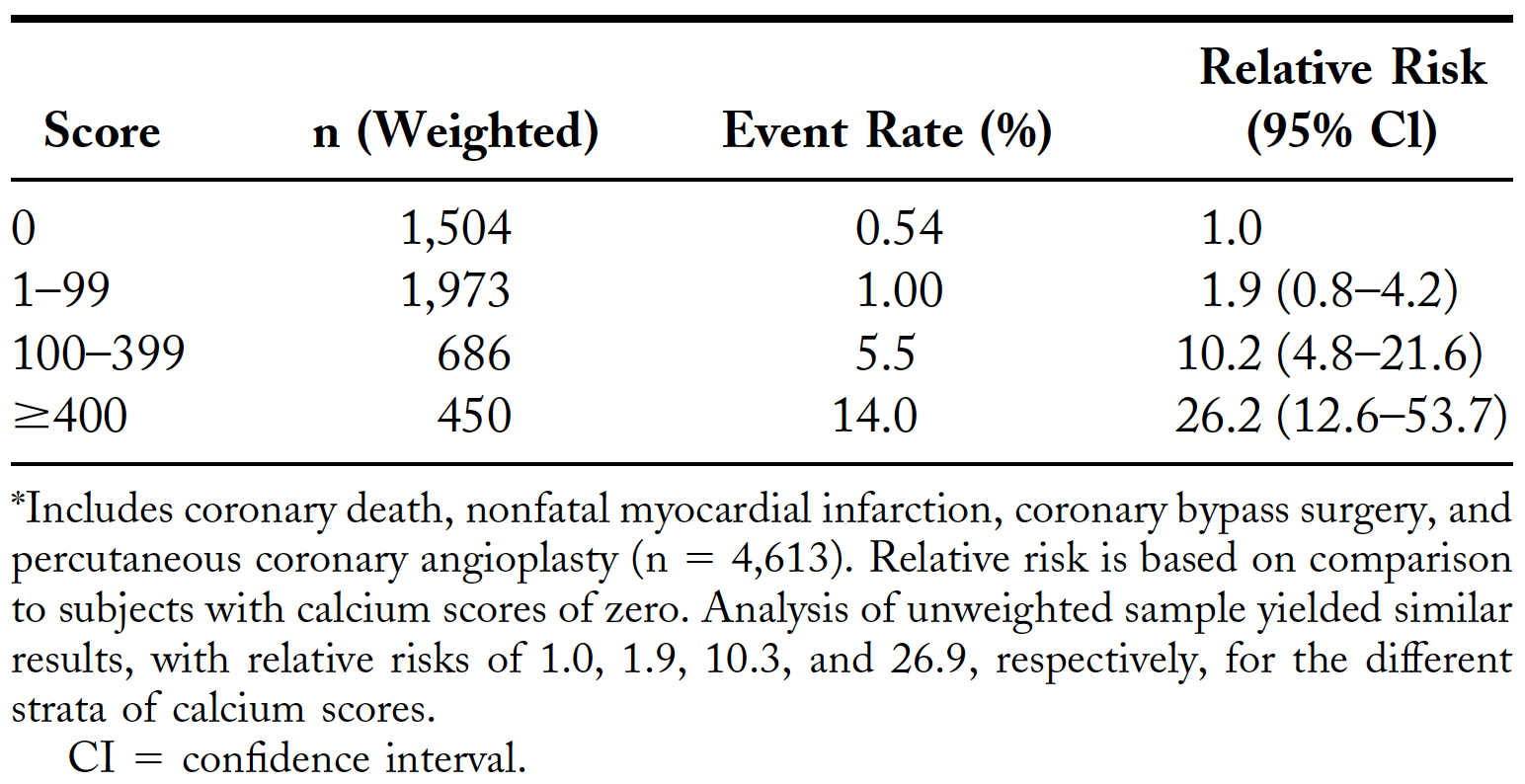
One study compared CAC with standard coronary heart disease risk factors (e.g., LDL cholesterol, HDL cholesterol, hypertension, current smoking, triglycerides) for predicting CVD events in 4,903 asymptomatic men and women between the ages of 50-70. They found that, after 4.3 years of follow-up, for CAC scores 100 or greater compared to scores below 100, the relative risk was 9.6 for all CVD events (95% CI, 6.7-13.9), 11.1 for all CHD events (95% CI, 7.3-16.7), and 9.2 for non-fatal heart attacks and death (95% CI, 4.9-17.3). The CAC score predicted events independently of standard risk factors and was better than the Framingham risk score in the prediction of events. To get a sense of the absolute risk, take a look at the event rates in Table 2 of the paper.
Table 2. CAC scores and all coronary disease events.*
In another study analyzing outcome data from the Multi-Ethnic Study of Atherosclerosis, investigators found that 10-year event rates for individuals between the ages of 55-64 with a CAC score of zero were 3.1% compared to 16.7% for people with a CAC score 300 or above.
A CAC score of zero may be the best predictor we have of low estimated CVD risk, but it does not grant one cardiac immortality, as you can see from these studies. There are a number of modifiable risk factors that should not be ignored because of a score of zero, for example, managing blood pressure, lipoproteins, insulin, smoking status, inflammation, glucose, stress, exercise, and sleep may all play a role in disease progression.
How do I use a CAC scan in my practice? Basically, I find it helpful in two scenarios (and this is discussed in great length in my interview with Dr. Ethan Weiss):
- A zero score in someone very “old” who is otherwise low risk and who has a low appetite for pharmacologic primary prevention, and
- A non-zero score in someone young (i.e., ~50 or younger) who is otherwise reluctant to engage in pharmacologic primary prevention.
A zero score in a 40-year-old only tells me they are “age-appropriate”—not at all that they are immune to atherosclerosis.
I’ll take a CAC of zero over non-zero all day long, make no mistake about it. If you only concern yourself with a 10-year risk horizon, it’s a mighty fine tool. But if you want to think about risk beyond that, you need to concern yourself not only with how many times you’ve had a break-in, but also the risk posed by the neighborhood you live in.
§
Thought experiment: in the next ten years, Harry’s going to fly for 10 hours each year for a total of 100 hours. This puts him in the lowest risk percentile for the risk of dying in a plane crash. Harry’s risk is 3%. His brother Steve flies more often and has a 10-year risk of 9%. Steve is probably feeling less confident about flying than his brother, but how confident is Harry? Does he feel like he’s free and clear from dying in a plane accident? If you thought your risk of death from flying in the next 10 years was 3%, would you change your travel habits?
Here’s the catch, those numbers do not represent anyone’s risk of death from plane crashes, but for the average individual who is 55-74-years-old with a CAC score of zero, he or she has a 3% chance of a “hard” atherosclerotic event like a heart attack, stroke, or cardiac death, according to the population in the Multi-Ethnic Study of Atherosclerosis. And for the same man or woman in the same age range with a score of 101-300, the risk is about 9%.
This isn’t a perfect analogy, but in both scenarios, I’m not exactly feeling immortal looking at a 3% risk on a 10-year horizon. If there are steps that I can take to lower my risk from 3% to 2%, I want to know about them, and after weighing the risks and benefits of each step, I may want to take them.
Let’s consider statins and another, newer lipid-lowering therapy, Proprotein Convertase Subtilisin/Kexin type 9 inhibitors (PCSK9i), and examine how they relate to CAC progression. (We tend to talk about drugs in this context, versus non-drug treatments, because so much of the data on the prevention of atherosclerotic cardiovascular disease (ASCVD) comes from drug trials, not because drugs are the only way to reduce the risk of heart disease.)
The latest guidelines from the American Heart Association (AHA) and the American College of Cardiology (ACC) on the primary prevention of ASCVD suggest using the ASCVD Risk Estimator Plus to estimate 10-year ASCVD risk for asymptomatic adults aged 40-79 years. Adults are then categorized into low (<5%), borderline (5 to <7.5%), intermediate (≥7.5 to <20%), or high (≥20%) 10-year risk.
For people in the borderline or intermediate predicted risk categories above, the ACC and AHA suggest the use of coronary artery calcium (CAC) scans to help refine the risk assessment. A higher CAC score, particularly when it’s in the 75th to 100th age/sex/race percentile,1You can find a risk score calculator and reference values here. can reclassify risk upward. On the other hand, a CAC score of zero can reclassify risk downward. The ACC notes that if CAC = 0 for individuals with an intermediate risk (≥7.5-20%) estimate, these people can avoid statins if they don’t have any higher-risk conditions (e.g., type 2 diabetes, family history of premature coronary heart disease, cigarette smoking) and it’s suggested they repeat the CAC scan in 5-10 years. If CAC is greater than 0, it’s deemed reasonable to use statins2See this summary on “Statin Treatment Recommendations” for more info. to benefit individuals and lower future risk of ASCVD.
Here’s where it gets complicated once you’re treated with statins. Statin therapy, demonstrated to reduce the clinical manifestation of ASCVD, can lead to an increase in CAC, yet equally potent low-density lipoprotein (LDL) lowering PCSK9i, which also reduce clinical events, do not. Why do two potent LDL lowering drug therapies, specifically statins and PCSK9i, have differing effects on CAC?
While we have a good understanding of the mechanisms behind how statins and PCSK9i work in the context of lowering LDL particles and LDL cholesterol, both drugs are known to have pleiotropic (i.e., multiple) effects. Statins directly reduce cholesterol synthesis and indirectly increase LDL clearance by the liver. PCSK9i inhibit, you guessed it, PCSK9, which are chaperone proteins that essentially degrade LDL receptors (LDLR). By inhibiting this protein, more LDLR are recycled and used at the surface of the cell, leading to greater LDL clearance by cells (particularly the liver) where LDLR are expressed.
While both drugs on their own can lower LDL-C in the range of a 50% reduction,3If statin and PCSK9i therapies are combined, as investigators did in these two studies, the reduction in LDL-C is additive, with patients sustaining levels in the range of 30-60 mg/dL. the drugs do this via different mechanisms. Researchers are hypothesizing that the PCSK9 protein, and therefore PCSK9i drugs, may have many effects beyond LDLR, although the hypotheses are somewhat preliminary. Understandable since PCSK9 was virtually unheard of prior to 2003.
Statins have reported effects other than lowering LDL concentrations, which include increased nitric oxide bioavailability, improvement of endothelial dysfunction, antioxidant properties, inhibition of inflammatory responses, and stabilization of atherosclerotic plaques.
As noted above, statins are known to increase the calcification of atherosclerotic plaque. Researchers have hypothesized that statins can promote or increase calcification through various actions.
If you want to get in the weeds (feel free to skip this section if you’re not on nerd alert):
Statins reduce cholesterol synthesis by inhibiting HMG-CoA reductase, which converts HMG-CoA to mevalonic acid, early in the multistep synthesis pathway that also affects prenylation pathways such as geranylgeranylation and farnesylation which affect (1) posttranslational protein modifications which in turn functions to anchor proteins to cell membranes and (2) GTPase molecules like Rho and Ras which influence the action of nuclear transcription factors critical to several cellular actions.
Researchers have hypothesized that statins (1) polarize macrophages to an M2, macrocalcification progressive phenotype, (2) inhibit vascular smooth muscle cell metalloproteinase secretion, (3) affect the fibrinolytic balance of vascular cells, and (4) affect the regulation of vascular smooth muscle cells inflammatory transcription factors. All of these may promote calcification. PCSK9i, which do not affect cholesterol synthesis or prenylation pathways, are not known to do any of the above.
PCSK9i, conversely, had no effect, up or down, on plaque calcification. One speculative reason why is that PCSK9i are more effective at lowering lipoprotein(a), or Lp(a) for short, than statins, and Lp(a) is positively correlated with calcification and an independent risk factor for aortic valve stenosis and calcification. In some patients, statins may increase Lp(a) concentration which hypothetically may also play a role in statin-induced calcification.
In light of what we’ve been discussing, calcification and CAC scores as a predictor of ASCVD, what are we to make of changes in the calcium content of coronary arteries brought on by statins? Here you have an intervention that has been shown to reduce CV events and yet may increase CAC. This presents a problem for evaluating the efficacy of lipid-lowering therapies using CAC scans. For example, should a statin-treated patient, with a CAC score of 200, whose apolipoprotein B levels, blood pressure, insulin, glucose, and other treatable risk factors have been adequately improved, worry if CAC increases over the course of 10 years?
In a 2018 study, investigators identified people without pre-existing ASCVD who got a CAC scan between 2002-2009. They followed these 13,644 patients (mean age of 50; 71% men) for an average of 9.4 years and sought to determine whether CAC can identify patients most likely to benefit from statin treatment. Comparing patients taking statins to those that didn’t, statin therapy was associated with reduced risk of major adverse cardiovascular events (MACE), in patients with CAC (i.e., a score greater than 0) (adjusted subhazard ratio: 0.76; 95% CI: 0.60-0.95; p = 0.015), but not in patients without CAC (i.e., CAC = 0) (adjusted subhazard ratio: 1.00; 95% CI: 0.79-1.27; p = 0.99). In other words, patients with a positive CAC benefited over ~10 years by taking a statin, but those with a CAC of zero did not benefit over this period of (relatively short) timeframe.
While the beneficial effects of statins on coronary artery plaque progression4The four major components of arterial plaque are dense calcium, necrotic core, fibro-fatty, and fibrous tissue. and ASCVD outcomes have been shown, statins increase coronary artery calcification, which is referred to in the literature as the “statin paradox” or the “plaque paradox.” (I always think of the physicist Niels Bohr’s quote when seeing this word: “How wonderful that we have met with a paradox. Now we have some hope of making progress.”) It’s possible that the observation of the plaques getting smaller and more calcified represents a conversion to a more stable plaque that’s less likely to rupture, although there doesn’t seem to be enough data to date to confirm or refute this hypothesis.
For the statin-treated patient, it’s possible that the progression of CAC over time is due to the treatment, and may not necessarily be an indicator of increased risk. This is why it might not make sense to monitor CAC once you’re on therapy. (There is one interesting study to suggest there’s a camp of patients that can benefit from monitoring CAC while on statin therapy that we’ll need to explore another time.)
This gets back to the larger point. This isn’t just about statins. This isn’t just about CAC. If you’re looking at a 3% risk of dying in an airplane crash over the next 10 years, you can truly prevent it. Don’t fly. It’s not so simple with ASCVD. There are a lot more dos and don’ts, and a number of factors that are outside our control. We need to be more nuanced, vigilant, and thoughtful in our approach. The fact that statins increasing coronary calcification may mislead us into thinking a patient isn’t benefiting from the therapy, when in fact he is after a follow-up CAC scan, is a good example of one of the ways in which we might be fooled. Biomarkers such as apoB (or LDL-P), LDL-C, non-HDL-C, CRP, and many others are important for risk assessment. If we want to delay the onset of the most inevitable disease to our species, lipid-lowering therapies and CAC scans are great tools to have if we use and interpret their effects appropriately, but we must not forget there are more things we need in our toolkit if we want to further improve our management, risk assessment, and risk modulation of ASCVD. And we’ll save that important topic for another day.
– Peter