Note: This narrative glossary is provided by Thomas Dayspring, M.D., FACP, FNLA, intended to accompany the series of podcast episodes between October 15 – October 19, 2018.
Lipids
Organic non-water soluble molecules (hydrophobic) as opposed to water (polar or hydrophilic). Some moieties are amphiphiles or amphiphilic: different ends of the molecule vary in polarity.
- Alkanes – flat, non-aromatic molecule with no double bonds
- Alkenes have a double bond
- Aromatic signifies ringed structures
Cellular structures (non-nuclear) important to lipids: ER, Golgi, endosomes (lysosomes), peroxisomes, membrane receptors, chaperone proteins.
Major classes of lipids
Steranes (no double bonds in rings), free sterols (unesterified), esterified sterols, stanols, steroids
- Free cholesterol (FC) has a hydroxy group (-OH) attached to carbon #3
- Cholesteryl ester (CE) has a Fatty Acid (FA) attached (esterified) to carbon #3
- Most common is oleic acid – hence the name cholesteryl oleate
Glycerolipids (Glycerides):
- Monoacylglycerol (MAG)-, Diacyl– (DAG), Triacyl-, TG, which respectively have one, two or three fatty acid acyl groups esterified (attached to) glycerol
- Phospholipids (PL) and lysophospholipids: Have a head group containing phosphorus, plus two or one FA acyl group (respectively) attached to glycerol
Fatty acid nomenclature:
- Short chain (< 5 carbons)
- Short: acetate, propionate, and butyrate
- Medium chain (6-12)
- Medium: lauric acid, caprylic acid
- Long chain (> 12)
- Long: oleic acid, stearic acid
Saturated, MUFA, PUFA, Trans
- The carbon atoms in a FA are numbered (n#). The last carbon in a chain is often called omega, and the first carbon alpha (nomenclature uses Greek alphabet)
- The location of a double bond between 2 carbon atoms is labeled as delta (∆)
- Cell and lipoprotein membrane structure consist of phospholipids and free cholesterol
- Lipid rafts are specifically composed membrane areas where so much signaling occurs
Structure of a lipoprotein
- Unilayer surface of amphiphilic phospholipids and free cholesterol
- Core a mixture of cholesteryl ester and TG
- Surface contains apolipoproteins which serve both structural and functional properties
- All apolipoproteins are transferable (exchangeable) except apolipoprotein B
Lipidome: term used to refer to all lipid components of lipoproteins
Proteome: term used to refer to all protein components of lipoproteins
- ApoB 100 and 48
- ApoA family: A-I, A-II, A-IV, A-V
- C-I, C-II, C-III (very important)
- ApoE
- Apo(a)
- Several others: eg D, F, H, J, K, L, M
- Phospholipid transfer protein
Lipoprotein Nomenclature: several classifications depending upon separation technique
- ApoB family (Lp-B)
- HDL or ApoA-I family (Lp-A)
Lipoprotein synthesis:
- apoB100 family (VLDL, IDL, LDL) made in liver
- apoB48 family (chylomicron) made in small intestine
- apoA-I made and secreted by liver & small intestine and assembled in plasma
Function of lipoproteins: lipid trafficking
- ApoB family: phospholipids and TG
- ApoA-I family: cholesterol and proteins
Reverse Cholesterol Transport concepts:
- TOTAL RCT = Direct RCT + Indirect RCT + Macrophage RCT + TICE
- TICE is transintestinal cholesterol efflux (enterocyte effluxes cholesterol to gut lumen)
- Macrophage RCT = efflux of cholesterol from arterial foam cells
Lipid Transfer Proteins:
- CETP (apoD) – mediates exchange of core particle lipids (CE and TG) between apoA-I or apoB particles (homotypic exchange) or between apoB & apoA-I family (heterotypic exchange)
- Lipid Inhibitory Transfer Factor (apoF) inhibits CETP-mediated exchange between apoB & apoA-I
Cellular Lipid transfer proteins:
- ATP Binding Cassette Transporters (ABCA1, ABCG1)– efflux free cholesterol out of cells to cholesterol acceptor proteins (usually apoA-I); ABCA1 to poorly lipidated (small HDL) and ABCG1 to larger, more mature HDLs
- ABCG5 and ABCG8 (sometimes called sterolin) – efflux unesterified (free)sterols from enterocytes to gut lumen or from hepatocytes to bile – loss of function increases sterol absorption – severe loss of function causes a type of FH called phytosterolemia
- ABCG4 – Effluxes phospholipids from hepatocytes to bile
- ABCB11– effluxes bile acids from hepatocytes into biliary system
- SR-B1 – (Scavenger receptor B1) effluxes or internalizes cholesteryl ester into or out of cells
- NPC1 or NPC2 – Niemann-Pick C1 – intracellular cholesterol transport protein
- NPC1L1 – Niemann-Pick C1–Like 1 protein – internalizes sterols into enterocytes from gut lumen or into hepatocytes from bile
- CD-36 – Cluster of differentiation protein – internalizes fatty acids into cells
Cholesterol homeostasis: a balance between synthesis and absorption
Synthesis: 37 step process starting with acetate to acetyl CoA
- First series steps are linear elongation of molecules – to mevalonic acid to squalene
- Next set of steps is aromatizing the structure (first ringed structure is lanosterol)
- Lanosterol to cholesterol utilizes two pathways
- Kandutsch Russell Path – goes thru creation of lathosterol which becomes cholesterol
- Bloch Path – goes thru creation of desmosterol which becomes cholesterol
- Both desmosterol and lathosterol concentrations are easily measured and serve as biomarkers reflective of cholesterol synthesis
Absorption: depends on:
- The balance of internalization of sterols, predominantly cholesterol from gut lumen into enterocytes utilizing the NPC1L1 protein, and
- The efflux of sterols out of the enterocyte into the gut lumen via ABCG5/G8 transporters.
The interplay of NPC1L1 and ABCG5/G8 at the hepatobiliary surface also comes into the balance.
The intestinal pool of absorbable free cholesterol derives (80-85%) from hepatobiliary delivery of exogenously produced free cholesterol and (2) ingested cholesterol (15-20%) (note – any cholesteryl ester that is ingested must be de-esterified to free cholesterol by pancreatic produced lipases (cholesterolase) before becoming available for intestinal absorption. On average humans absorb ~54% of the gut pool of cholesterol. But depending on the function of NPC1L1 and ABCG5/G8, 20% are hyper-absorbers of sterols and 20% hypo-absorbers. Genetic loss/partial loss of function of NPC1L1 is associated with reduced LDL-C and significantly reduced risk of CHD. Note that ezetimibe interferes with the action of NPC1L1.
Because NPC1L1 has a much higher affinity for cholesterol than for phytosterols or any stanol, concentrations of phytosterols such as sitosterol and campesterol or a stanol like cholestanol are used clinically to reflect cholesterol absorption.
Once free cholesterol enters the enterocyte it can be effluxed to and incorporated into HDL particles via ABCA1 transporter or along with phospholipids, be incorporated into the surface of a chylomicron, effluxed back to the gut lumen via ABCG5/G8 transporters or be esterified via the action of acyl-cholesterol acyltransferase (ACAT) to form cholesteryl ester which joins with TG to form the core of the chylomicron. Note any phytosterols that made it into the enterocyte are normally effluxed out immediately by ABCG5/G8. Phytosterols are also poor substrates for esterification by ACAT and not likely to wind up in the core of a chylomicron. However unesterified phytosterols can be effluxed by intestinal ABCA1 to HDL surfaces.
Excretion: Cholesterol can be transformed into other molecules – steroid hormones (gonads, adrenal cortex) or bile acids (liver) but it cannot be metabolized (thus cannot serve as an energy substrate). Since cellular excess of cholesterol is toxic it has to be returned to the gut and excreted. Within the gut, cholesterol can be desaturated and converted by gut microbes into cholestanol or coprostanol (isoforms or molecular mirror images of one another). Since stanols like cholestanol cannot be absorbed, they are excreted.
Bile acids are synthesized from cholesterol in the liver and then enter the bile via ABCB11 transporters where they enter the intestine. Along with phospholipids, which emulsify gut lipids (fatty acids and free cholesterol or other sterols), bile acids create biliary micelles (collections of lipids) which deliver lipids to the intestinal villi, where they can be absorbed.
Dietary TG and phospholipids are much too large to be absorbed by the intestine. Thus, they must be respectively de-esterified by pancreatic lipases into fatty acids or monoacylglycerols (or a lysophospholipid) before they can be absorbed. Once the FA are absorbed they are re-esterified to glycerol forming new triacylglycerols or phospholipids, which can be incorporated into chylomicrons.
Lipolysis: Any esterified lipid, within a cell or within a lipoprotein can be de-esterified (called hydrolysis) via the action of enzymes called lipases.
Lipoprotein lipase (LPL) is a triglyceridase which when activated by the ligand apoC-II (of which chylomicrons and VLDLs carry several copies of) hydrolyzes TG to FA or MAGs. It is advanced lipidology, but also critical to lipolysis of chylos (chylomicrons) and VLDL are apoA-V and myocyte or adipocyte endothelial expression of both glycosylphosphatidylinositol HDL BP 1 (GPIHBP1) and lipase maturation factor (LMF). TG-rich lipoprotein lipolysis is inhibited by apoC-III.
Hepatic lipase (HL) is a triglyceridase/phospholipase that hydrolyses esterified lipids on smaller lipoproteins (IDL, LDL, and HDL).
Endothelial lipase (EL) is a phospholipase that hydrolyzes PL on very small lipoproteins (HDL).
Multiple other types of phospholipases exist.
Lipoprotein clearance
Apart from lipoprotein catabolism by lipolysis, particles can be cleared from plasma via several cellular membrane receptors. The apoB100 particles can be cleared by the LDL receptor (LDLR) which although ubiquitous, are most heavily expressed in the liver. The LDLR binds to a specific section (called the LDL receptor binding domain) on apoB100. ApoB48 does not have that domain and thus does not bind to LDLR.
ApoE is also a ligand for the LDLR (explaining the rapid clearance of VLDLs and IDLs) and for a similar receptor called the LDL receptor-related protein (LRP) which clears apoE-containing lipoproteins. LDL clearance is much slower than chylos, VLDLs, and IDLs as LDL, as LDL typically only has apoB100 (and no apoE). LDLs which do carry apoE are rapidly cleared.
ApoC-III blocks clearance of apoB100 particles explaining the longer half-life of pathological remnant lipoproteins and any LDL that might carry apoC-III. HDLs are typically delipidated by cell surface receptors (SR-B1) but can be internalized at the liver by holoparticle receptors. Oxidized apoB-containing lipoproteins are rapidly cleared from plasma by hepatic scavenger receptors explaining the minimal exitance (concentrations) of oxidized LDL in plasma.
Lipid/lipoprotein metrics
Plasma volumes are measured as liters (L) or tenths of a liter called deciliters (dL) or thousandths of a liter called milliliters (mL). For reference note that one ounce is 30 mL, a teaspoon is 5 mL and a tablespoon 15 ml (half an ounce).
Weights of molecules are usually reported in grams (g) milligrams (mg) or a term called moles (mols) or fractions of a mole: a billionth of a mole is called a nanomole (nmol). Moles represent the # of atoms, molecules, particles.
Cholesterol metrics
Total Cholesterol (TC): the cholesterol carried in all of the lipoproteins per dL of plasma.
HDL-C: The cholesterol content within all of the HDL particles per dL of plasma.
LDL-C Calculated: The cholesterol content within all of the LDL and IDL and Lp(a) particles per dL of plasma.
LDL-C = TC – [HDL-C + VLDL-C] where VLDL-C is estimated by dividing TG/5
Measured: a lab assay that measures sterols within the LDLs and Lp(a) particles per dL. IDL-C is not a component.
VLDL-C: The cholesterol content within all of the VLDL particles per dL of plasma. It is calculated by TG/5 (on the assumption that when fasting all TG are in VLDL and a normally composed VLDL carries 5 x more TG than cholesterol. The calculation is not the most reliable way of evaluating VLDL-C especially as TG levels rise. VLDL-C can also be estimated by subtracting directly measured (not calculated) LDL-C and HDL-C from TC.
VLDL-C = TC – [LDL-C + HDL-C]
Although VLDL-C may have some correlation with atherogenic remnants, it should be used with caution as a surrogate of remnant-cholesterol because not all VLDLs are necessarily pathologic remnants.
Non-HDL-C: A calculation that represents the cholesterol content within the apoB-lipoproteins (which obviously are not HDL particles) in a dL of plasma. It is the cholesterol within any chylomicrons, VLDLs, IDLs, LDLs and Lp(a) particles per dL. Since apoB-cholesterol is potentially atherogenic cholesterol Non-HDL-C is used as a calculated surrogate of apoB particles.
Non-HDL-C = TC – HDL-C
TG metrics
Recall that TG consists of a glycerol backbone esterified (attached) to 3 fatty acid acyl groups. Because the FA composition of each TG molecule can vary so widely and each TG can have a different or the same FA at each of the 3 carbons of glycerol (for nomenclature purposes each carbon of glycerol is designated as a stereospecific binding number: sn1, sn2, and sn3). Thus there are numerous potential TG species, each of with a different molecular weight.
To determine TG concentrations, labs add a plant lipoprotein lipase to the serum de-esterify the FA. The glycerol is then measured – since every TG molecule has one glycerol – it is easy to translate mols glycerol to mols of TG. So in essence labs measure glycerol, not TG. Most humans have little free glycerol in plasma, but for those who do (a glycerol kinase deficiency or using too much exogenous glycerol) erroneous hypertriglyceridemia will be reported (called pseudohypertriglyceridemia). True Health Diagnostics is one of the few labs that checks glycerol levels in anyone with a TG ≥ 400 mg/dL. This is called glycerol blanking. Note, no phospholipase is added to the serum, so phospholipids will not influence glycerol (TG) measurements.
Subparticle cholesterol metrics
HDL3-C: the cholesterol content of the smaller HDL species. Due to their small size smaller HDLs traffic considerably less cholesterol than do larger species.
HDL2-C: the cholesterol content of the large HDL species. Since the large HDLs compared to small, traffic most of the cholesterol with HDLs, most persons with an HDL-C < 40 mg/dL will have low HDL2-C. By far the most common clinical scenario where this occurs is insulin resistance (IR) and rising TG levels. Thus, measurement of HDL2-C is used as a lipid concentration surrogate of insulin resistance, prediabetes or T2 diabetes.
Total HDL-C = HDL1-C + HDL2-C +HDL3-C
Most persons have no appreciable amount of the very large HDL1 particles so for practical circumstances Total HDL-C = HDL2-C + HDL3-C. Labs measure Total HDL-C and HDL3-C and then calculate HDL2-C using the formula HDL2-C = Total HDL-C – HDL3-C.
Of interest is that because the volume of a spherical lipoprotein is a 3rd power of the radius, it is the larger HDL2-C that drives HDL-C, especially when concentrations are > 40 mg/dL. However what drives HDL-P are the smaller, cholesterol-poor HDL species. Size of an HDL particle has no relationship to HDL function or reverse cholesterol transport.
Subparticle TG – LDL-TG, HDL-TG – is not routinely measured outside of research studies.
TG-rich HDLs and LDLs, most commonly seen in IR (insulin-resistant) states, are pathological particles prone to lipolysis – this leads to discordance between particle cholesterol and particle number metrics. TG-rich HDLs are typically dysfunctional. TG-rich LDLs usually undergo lipolysis (vis hepatic lipase) creating smaller, less efficiently cleared LDL species further leading to significant increases in small and total LDL-P (apoB).
Lipoprotein particle concentrations can be measured by several techniques, but the one with by far the most published CV data are those determined by Nuclear Magnetic Resonance Spectroscopy (NMR) – analysis of the spectral signals generated by methyl groups on lipids within lipoproteins is used to generate particle sizes, particle concentrations, and particle lipid concentrations.
Commercially NMR spectroscopy can report total VLDL-P, LDL-P, small LDL-P, HDL-P.
Concentrations are reported as nmol/L (the apoB particles) and µmol/L for HDLs, which makes it obvious there are way more HDL particles than apoB particles in plasma.
An LDL particle count of 1000 nmol/L means that the number of low-density lipoprotein particles circulating in a liter of plasma is 1000 nanomoles. It does not mean there are 1000 particles per liter. There are actually quintillions of LDL particles circulating in plasma.
Presence of large VLDL-P is highly suspicious of insulin resistance (IR). Normal people make few if any large VLDL particles. Typically, large VLDLs are seen with increasing TG.
Likewise, the presence of small LDL or HDL size or increased small LDL-P or reduced HDL-P are lipoprotein abnormalities suggestive of IR.
The only NMR metric that has been validated as a goal of therapy is LDL-P. Since total LDL-P = Large LDL-P + small LDL-P, total LDL-P is driven by small LDL-P. Note that Lp(a) particles, if they exist, are counted as LDLs.
Because they contain very few lipids, NMR cannot measure pre-beta HDLs, but they represent < 5% of total HDL particles and their measurement serves no clinical purposes.
NMR cannot differentiate any protein such as apoprotein(a), so NMR cannot be used to measure Lp(a) concentrations. Total LDL-P is the sum of LDL particles that do not carry apoprotein (a) + any LDLs that do.
Total LDL-P = non-apo(a)LDL-P + Lp(a)-P
Important because statins lower non-apo(a)LDL-P, but not Lp(a)-P, but one still gets the total LDL-P benefit.
Apolipoproteins B: There is one molecule of apolipoprotein B on every plasma lipoprotein that is not an HDL, or a particle known as Lp(x). Thus, there is an apoB on all VLDLs, IDLs, and LDLs (and any Lp(a) particles that may exist), although because of plasma residence times 90-95% of the apoB particles are LDLs. When one measures apoB, its only use is as a biomarker indicative of potentially atherogenic lipoproteins, the vast majority of which are LDLs. ApoB tells one little about VLDL or remnant concentrations.
Apolipoprotein A-I: there are from 1-5 molecules of apoA-I per HDL particle so apoA-I serves as an estimation of HDL particle count. ApoA-I is best used as a marker of CV risk in drug naive individuals. Since changing apoA-I therapeutically has never been demonstrated to be associated with reduced CV events, neither apoA-I or any ratio incorporating it can be used as a goal of therapy.
Lipoprotein sizes: All lipoprotein subclasses exist in a heterogeneous mixture of diameters and can be classified as large, intermediate size or small. With respect to LDLs, LDL receptors more easily recognize, and clear, intermediate sized LDLs because on large LDLs or small LDLs, the LDL binding receptor domain on apoB may be distorted. Thus, large and small LDL particle have delayed clearance. Any lipoprotein < 70 nm in diameter can pass thru endothelial barriers and enter the arterial wall. The largest LDL is ~26 nm in diameter. Mid- and smaller VLDLs and chylomicron remnants and IDLs are all < 70nm in diameter.
Lipoprotein (a): is an LDL-like particle to which 1 molecule of apoprotein (a) has bonded (to apoB).
Plasminogen: a protein involved with thrombolysis, like many proteins involved with coagulation, consists of a series of looped-amino acids called kringles. Plasminogen has five kringles (numbered I-V) attached to an active protease (protein lysing enzyme) domain (segment).
Apoprotein (a): produced in the liver has two kringles numbered IV and V. Kringle IV in apoprotein (a), unlike KIV in plasminogen, has ten subsegments of which sub-segment 2 (KIV-2) can have a very variable number of amino acids. Thus the molecular weight or size of apo(a) depends on KIV-2.
Hepatocytes can produce smaller (lower MW) apo(a) peptides much quicker than they can larger (higher MW) isoforms. Thus, persons who produce small apo(a) tend to have much higher Lp(a) particle counts [Lp(a)-P], compared to those producing high MW apo(a).
Lp(a) Metrics
Lp(a)-C: refers to the cholesterol within all of the Lp(a) particles the exit in a dL of plasma. There is no commercially available test, including that measured by vertical ultracentrifugation that accurately measures Lp(a)-C.
Lp(a) mass: the mass of every molecule within all of the Lp(a) particles per dL. It is the mass of apo(a), apoB100, any other apoproteins, and every lipid molecule, including TG, free and esterified cholesterol, and phospholipids.
Lp(a)-P: Because there is no MW of these macromolecular structures, Lp(a) in mg/dL cannot be accurately converted to nmol/L.
Lp(a)-P – offered at True Health Diagnostics – it is the nmol/L concentration of Lp(a) particles. Lipoproteins are separated in an electrophoretic gel and the Lp(a) segment is immune-stained with apo-B. That can be transformed into Lp(a)-apoB which because there is one apoB per Lp(a) particle makes the test an Lp(a) particle number.
Remnant metrics
Remnant lipoproteins are potentially atherogenic smaller VLDL and chylomicron particles that have become cholesterol-rich via lipolysis and CETP activity. Not all medium or smaller sized VLDL particles are atherogenic as most carry multiple copies of apoE and are rapidly cleared by the various LDL receptors.
Large VLDL-P represents a large TG-rich VLDL and is not a remnant particle, but it might after lipolysis, become a remnant. Likewise, VLDL-C (calculated or measured) is not an accurate test of remnants as not every VLDL particle is atherogenic (as most are cleared by apoE-binding receptors).
Most VLDL remnants have apoC-III attached, but that cannot be measured. Many prefer to calculate VLDL-C by subtracting measured LDL-C and HDL-C from total cholesterol. But again, not every VLDL particle carrying cholesterol is arterial wall bound. If one uses VLDL-C in this manner, levels should not be higher than 15-20 mg/dL.
Lipid Pathology
Hyperbetalipoproteinemia: major atherosclerotic CV risk. (Too many apoB100-containing lipoproteins.)
FH and insulin resistance, but also seen in some after high-fat, ketotic diets.
Hypobetalipoproteinemia: Reduced numbers of apoB-containing lipoproteins,associated with reduced CV risk – defined as a drug-naive LDL-C < 50 mg/dL.
Abetalipoproteinemia: no apoB particles – associated with many morbidities.
Complex hypobetalipoproteinemia: characterized by low LDL-C and HDL-C.
Dysbetalipoproteinemia (formerly called Type III): a remnant disorder characterized by very high LDL-C and triglycerides but normal apoB and often have a zero LDL-P (NMR). The elevated TG and cholesterol are in VLDL and IDL particles. ApoE genotype cannot be used to make the diagnosis of Type III.
Xenosterolemia (Phytosterolemia): >99th percentile levels of phytosterols.
TG disorders: Levels > 1000 mg/dL are severe and > 2000 mg/dL very severe and may be associated with pancreatitis. Levels < 1000 mg/dL are often related to insulin resistance & T2D (and very high apoB and LDL-P).
HDL disorders: Hyperalphalipoproteinemia (HDL-C > 60-70 mg/dL) and hypoalphalipoproteinemia (HDL-C < 20 mg/dL).
Cholesterol-rich HDLs may be dysfunctional. Dividing HDL-C (in molar concentration) by HDL-P will provide how many cholesterol molecules are in the HDL particles. Normal is 43 – Levels > 53 are abnormal.
Lipid modulating drugs
Upregulation of LDL receptors which enhances apoB-particle clearance.
Bile acid sequestrants: cholestyramine or colesevelam. These prevent reabsorption of bile acids forcing liver to deplete its cholesterol pool to synthesize new BA. Leads to LDLR upregulation. Administration of and compliance with these meds is difficult. Colesevelam also approved to lower glucose.
Statins: hydrophilic and lipophilic types – apoB-lowering potency varies per statin and per dose. Inhibit HMG CoA reductase the rate-limiting enzyme in the cholesterol synthesis pathway. All statins have clinical outcome trial data. First to market in 1987 was lovastatin, followed by pravastatin and simvastatin. First outcome trial ever published was in 1994 4S with simvastatin followed soon by a primary prevention trial using pravastatin (WOSCOPs). Mg per mg, pitavastatin is the most potent lowering statin and also has the fewest drug-drug interactions and least glucose-raising potential. Depending on dose and frequency of administration statins can lower apoB by 10-40%. Statins in some can cause a reflex hyperabsorption of cholesterol. Effect of statin on synthesis and absorption can be assessed by following the synthesis markers desmosterol and the absorption markers lathosterol as well as sitosterol, campesterol, and cholestanol. Statins can also paradoxically increase PCSK9 activity. Thus hypo-response to statins may be seen in hyper-absorbers of cholesterol.
Ezetimibe: blocks the action of the NPC1L1 proteins which is the sterol influx protein located at gut lumen/enterocyte interface and the hepatobiliary interface. Thu, ezetimibe has two paths which will reduce cholesterol pools in the liver, which upregulates LDLR. Also blocks internalization of phytosterols. In some ezetimibe monotherapy can cause a reflex hyper-synthesis of cholesterol and increased PCSK9 activity.
LDL receptor: PCSK9 inhibitors: PCSK9 is an enzyme that binds to LDL receptors and enhance their catabolism in lysosomes. Inhibitors increase the lifespan of LDL receptors and thus significantly enhance apoB particle clearance. Two monoclonal antibodies which bind to PCSK9 are available: alirocumab (Praluent) and evolocumab (Repatha). They are injected subcutaneously every 2-4 weeks. They can reduce apoB 20- 40%’ Although not indicated by the FDA, they can also reduce Lp(a) by 25-30%. So far no side effects of consequence, including any CNS symptoms, has been seen in clinical trials despite extreme ability to lower LDL metrics (especially LDL-C). 3rd party mayors mandate their use be limited to very high-risk individuals (have had a clinical event or FH) who have not been able to achieve LDL goals with maximally tolerated statin therapy.
TG-altering drugs: fibrates (2 exist in the US: gemfibrozil and fenofibrate (including a more bioavailable form called fenofibric acid). These are potent TG-lowering drugs that also are indicated to lower LDL-C. Their PPAR-alpha agonists MOA is complex but they inhibit DGAT (a key enzyme in TG synthesis) and reduce apoC-III and influence numerous cell membrane receptors like SRB1 and ABCA1. Fibrates have positive primary and secondary prevention trials (Helsinki and VA-HIT) and fenofibrate has been shown to reduce microvascular events: retinopathy, neuropathic ulcerations and distal amputations, gout and uric acid, proteinuria and improve eGFR. (FIELD and ACCORD trials). Although package inserts carry warnings, due to drug-drug interactions it is much safer to combine fenofibrate with statins than gemfibrozil which should not be used. Fenofibrate lowers LDL-P and is the best available drug at raising HDL-P.
Niacin: a drug which lowers apoB, Lp(a), TG and raises HDL-C but paradoxically not HDL-P. It has never reduced the primary endpoint in any clinical outcome trial. Its only positive data are small angiographic trials not empowered to evaluate outcomes. Niacin is well known to have many adverse clinical and metabolic effects as well as increases insulin resistance.
Omega-3 Fatty Acid Rx: currently have no published outcome data support, but reduce TG when used at dosages of 4 grams or higher. A clinical outcome trial (REDUCE-IT) which used high dose EPA, just reported a positive finding, and will be presented at the November 2018 AHA meeting. A similar trial using EPA plus DHA is in progress.
Phytosterols and stanols: By reducing cholesterol incorporation into biliary micelles can reduce cholesterol absorption – but in hyper-absorbers will increase plasma phytosterol levels (likely not desirable). Thus, use caution with their use in hyper-absorbers of cholesterol.
Advanced discussion: UNDERSTANDING STEROL & STANOL BIOMARKER TESTING
Sterols are organic ring-structured molecules each with a hydroxy group on the first ring at the 3rd carbon, a double bond in the second ring at the 5th carbon but differing and uniquely characterized by the side chains off of carbon 17 located in the last ring. Stanols are simply sterols whose second ring is saturated (no double bond).
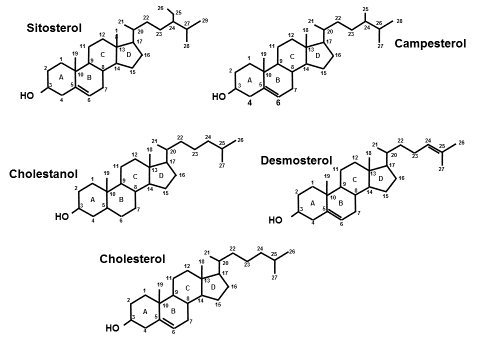
Unesterified or free cholesterol is a critically important molecule for humans yet in cellular excess is toxic so its homeostasis is tightly regulated by a balance of synthesis and absorption. Every cell can synthesize cholesterol in a complex, multistep process starting with acetate. The only way for the body to rid itself of cholesterol is to excrete it via the stool or convert it to other usable or excretable molecules such as steroids, cholestanol, and bile acids. Every cell in the body can both synthesize cholesterol as well as efflux excess quantities out to circulating apoA1 or apoA1-containing high-density lipoproteins (HDL). HDLs can directly traffic that cholesterol to steroidogenic tissues, adipocytes or the liver (direct reverse cholesterol transport) or transfer it to low-density lipoproteins who return it to the liver (indirect reverse cholesterol transport). Utilizing cellular membrane sterol efflux proteins [ATP-binding cassette transporters (ABCG5, ABCG8)] liver cells (hepatocytes) can excrete unneeded cholesterol into the biliary system and intestinal cells (enterocytes) into the intestinal lumen. Apart from cellular synthesis, part of cholesterol regulation is by utilizing membrane located sterol influx protein [Niemann Pick C1 like 1 (NPC1L1)] in enterocytes to absorb free cholesterol from the gut lumen and in hepatocytes to reabsorb cholesterol from the biliary pool. Elevated total and/or LDL-cholesterol provides no information on whether such concentrations might be related to hyper-synthesis, hyperabsorption, neither or both. Because they serve no physiologic role dietary neither phytosterols, phytostanols nor other stanols are normally absorbed.
In the cholesterol synthesis pathways, the penultimate sterols prior to cholesterol are desmosterol and lathosterol and both can serve as biomarkers to evaluate cholesterol synthesis. Phytosterols (e.g. sitosterol, campesterol) serve no physiologic role, cannot be synthesized in the body and have markedly reduced absorption capabilities compared with cholesterol. Likewise, all stanols including cholestanol, a gut microbial metabolite of cholesterol have extremely limited absorption. Thus sterol/stanols can serve as biomarkers reflective of cholesterol absorption. In a given individual the sterol-binding domain segments of NPC1L1 and ABCG5/G8 have very distinct affinities for cholesterol, and each of the phytosterols and stanols. This explains why cholesterol hyper-absorptive states may be associated with one, two or all three of the absorption biomarkers.
Serum concentrations of cholesterol absorption/synthesis biomarkers have been validated as tools for the diagnosis of genetic sterol disorders, the evaluation of cholesterol homeostasis, the evaluation of cardiovascular risk, and for choosing lipid-modulating therapies. Sterol biomarkers are reported in both absolute concentrations and as ratios to total cholesterol. The ratios can be difficult to interpret if the patient is on a cholesterol modulating drug and thus when making decisions in patients on such drugs, the absolute concentrations are best used. This also explains why although they usually correlate absolute concentration vs ratio to total cholesterol results may be discordant.
Hyperabsorption is an important and modifiable risk factor known to be associated with women ≥ age 50, and apoE4 alleles. As a general rule, hyper-absorbers of cholesterol tend to be hypo-synthesizers and vice versa and such a pattern is often associated with increased CV risk. The disease phytosterolemia is very rare and the sitosterol, campesterol levels will be in the 100-300 µg/mL range. Although elevations of phytosterols are associated with CV risk, one cannot state with assurance the phytosterols are causal of atherosclerosis. Phytosterols used as nutritional supplements to reduce LDL-cholesterol may significantly increase phytosterol levels in hyper-absorbers and should be avoided in those situations. Phytostanols such as sitostanol (Benecol®) cannot be absorbed to any appreciable degree and can be used in patients with hyperabsorption of sterols.
Hypo-absorption of cholesterol, genetic or drug-induced is desirable and associated with lower cardiovascular (CV) risk and as an isolated finding requires no treatment per se. Ezetimibe, fibrates, bile acid sequestrants (BAS), probiotics, berberine and esterified sitostanol (Benecol®) reduce cholesterol absorption. Statins decrease cholesterol synthesis but may increase absorption. Low desmosterol has been associated with cognitive impairment which raises the question of how much synthesis inhibition is desirable.
When LDL-P (apoB) are elevated in the face of normal sterol values, there may be a problem with overproduction (typically related to TG abnormalities) or decreased clearance of apoB particles (LDL receptor issues, defective apoB, PCSK9 gain of function issues).
With respect to treatment decisions in persons with sterol synthesis/absorption abnormalities pay attention to the following:
- Unless phytosterolemia is present or the patient is in the very high CV risk category, LDL-P (apoB) is the goal of therapy not the sterol levels per se. The higher the overall CV risk of the patient, the more aggressive is the apoB (LDL-P) goal of therapy;
- After lifestyle, unless TG are > 500 mg/dL statins are usually the first line LDL-P lowering therapy; and
- Depending on overall CV risk, if hyperabsorption is present in the face of elevated LDL-P, ezetimibe or low dose statin/ezetimibe is an effective therapy. However even with hypo-absorption, if apoB is elevated on statin monotherapy adding ezetimibe can still be effective. Although such use is off-label, ezetimibe, because of its long half-life can be used every other day.