Before I get into this post I want to lay a few things out.
- This post is written mostly for doctors, but also for patients who really want to understand this topic, if for no other reason than to help them choose the right doctors. I don’t go out of my way to simplify the terminology and I assume the reader is familiar with the topics covered in the cholesterol series I wrote. If you encounter a term you don’t understand, Google is a pretty good place to find the definition.
- I will not use this post to in any way get into prescriptive strategies, which involve modifications of nutrition, hormones, and yes, lots of drugs across four or five classes (i.e., much more than just statins) depending on the specific situation at hand and the risk appetite of the patient and physician, as well as the other comorbidities that must be co-managed. Even if I wanted to write out all of my prescriptive leanings I could not do it briefly.
- Please do not email your lab results or ask me to weigh in on your case. You know the disclaimer: I can’t practice medicine on a blog or over email.
There was a day when the only thing I argued about was who the greatest boxer of all time was. (I’m fighting all urges to turn this post into a manifesto of 1965-67 Muhammad Ali vs. 1938-41 Joe Louis vs. 1940’s Sugar Ray Robinson vs. 1937-42 Henry Armstrong.)
Today, however, I find myself arguing about so many things—some of them actually important—from why symptomatic women should receive hormone replacement therapy after menopause (and, by extension, why the Women’s Health Initiative tells us so, if you know how to read it) to why monotherapy with T4 for hypothyroidism is a recipe for disaster for most patients. But there is nothing I find myself debating more than the misconceptions most doctors have about heart disease. This is especially troubling since heart disease kills more Americans than any other disease. To put this in perspective, a woman in the United States is 7 to 8 times more likely to die from heart disease than she is from breast cancer.
Here are the typical arguments put forth, almost always by doctors, which invariably result in my need/desire to counter:
- Heart disease is caused by too much “bad” cholesterol (LDL-C).
- LDL-C is the only target of therapy you need to worry about.
- Calcium scores and CT angiograms (CTA) are great ways to further risk-stratify (the corollary: when these tests are negative, there is no need to treat the patient).
- Atherosclerosis is a “pipe narrowing” disease (ok, nobody uses these words, but they imply this by saying it’s a luminal narrowing disease).
- There is no role for preventatively treating young people, except in very rare cases like familial hypercholesterolemia.
Briefly, here are my counters:
- Atherosclerosis is caused by an inflammatory response to sterols in artery walls. Sterol delivery is lipoprotein-mediated, and therefore much better predicted by the number of lipoprotein particles (LDL-P) than by the cholesterol they carry (LDL-C) [Bonus point: always measure Lp(a)-P in your patients—but we’ll have to save Lp(a) for another day; it certainly owns its own blog post].
- Ditto point #1. And don’t ever bring up LDL-C again.
- Calcium scores and CT angiograms of exceptional quality (the operative word being exceptional—most are not) are helpful in a few settings, but this assertion is patently false, and I will leave this discussion for another blog post as the topic is too rich in nuance for a few lines.
- We’ll discuss this today.
- By necessity, we’ll be forced to confront this today, also.
Before diving into this topic it’s really important for me to acknowledge the person who has taught me almost everything I know about this disease, beginning back in 2011 when I first became aware that I basically had no idea what atherosclerosis was. For many years Dr. Tom Dayspring’s generosity has been remarkable and I’m humbled to be his most sponge-like student. Tom has not only given me an on-the-side lipidology fellowship, but he has also introduced me to the finest lipidologists and cardiologists in the country who have, in turn, been incredibly generous with their time and knowledge. I’m not the only one to benefit from Tom’s wisdom and generosity. I had dinner with Tom’s son and his wife once and I described Tom to them as a national treasure. That’s really how I feel about him. He is a nationally-recognized educator and his writing and presentations are devoured by fanatics like me across the globe. With Tom’s permission, I’ve deconstructed a video he put together into a series of figures which I’ll use to begin this discussion of how atherosclerosis actually takes place.
The physics of luminal narrowing
Traditionally, the atherosclerotic process was believed to involve plaque accumulation that prompted the gradual narrowing of the lumen, with the eventual development of stenosis. Stenosis then caused impaired control of flow (stable angina) and plaque rupture and thrombosis (unstable angina and MI). Consequently, prevailing opinion held that coronary angiography would be able to gauge the atherosclerotic process at all stages of disease.
However, in 1987, Glagov and colleagues proposed an alternative model of atherosclerosis development. After performing histological analyses of coronary artery sections, Glagov et al. reported that early atherosclerosis was characterized by plaque accumulation in the vessel wall and enlargement of the external elastic membrane (EEM) without a change in lumen size.
As atherosclerosis progressed, they found that plaque continued to accumulate in the vessel wall until the lesion occupied approximately 40% of the area within the EEM. At this point, the lumen area began to narrow. These findings have since been confirmed by intravascular ultrasound (IVUS). Due to the complex remodeling that occurs in the earlier stages of atherosclerosis, coronary angiography, which only visualizes the lumen, tends to underestimate the degree of atherosclerosis. In other words, atherosclerosis is well under way long before angiography is able to identify it.
I was reminded of the words of my Pathology professor back in the first year of medical school, “The only doctors who actually understand atherosclerosis are pathologists.” I would add lipidologists to that list, but I saw his point.
Most people, doctors included, think atherosclerosis is a luminal-narrowing condition—a so-called “pipe narrowing” condition. It’s true that eventually the lumen of a diseased vessel does narrow, but this is sort of like saying the defining feature of a subprime collateralized debt obligation (CDO) is the inevitable default on its underlying assets. By the time that happens, eleven other pathologic things have already happened and you’ve missed the opportunity for the most impactful intervention to prevent the cascade of events from occurring at all.
To reiterate: atherosclerosis development begins with plaque accumulation in the vessel wall, which is accompanied by expansion of the outer vessel wall without a change in the size of the lumen. Only in advanced disease, and after significant plaque accumulation, does the lumen narrow.
Michael Rothberg wrote a fantastic article on the misconception of the “clogged pipe” model of atherosclerosis. He opens with the following story:
A recent advertisement on the back cover of a special health issue of the New York Times Magazine section read “Ironic that a plumber came to us to help him remove a clog.” The ad referred to doctors in the cardiac catheterization laboratory as “one kind of pipe specialist,” and noted that the patient in the ad returned to work “just 2 days after having his own pipes cleaned out.” Although the image of coronary arteries as kitchen pipes clogged with fat is simple, familiar, and evocative, it is also wrong [emphasis mine].
Dr. Rothberg goes on to explain that for patients with stable disease, local interventions can only relieve symptoms; they do not prevent future myocardial infarctions. To be clear, at least 12 randomized trials conducted between 1987 and 2007, involving more than 5,000 patients, have found no reduction in myocardial infarction attributable to angioplasty in any of its forms. And yet, despite this overwhelming evidence, the plumbing model, complete with blockages that can be fixed, continues to be used to explain stable coronary disease to patients, who understandably assume that angioplasty or stents will prevent heart attacks—which they patently do not.
The root of the problem, in my view at least, is that we as doctors—and by extension, our patients and media—spend too much time looking at images like this (angiograms of coronary arteries complete with “clogged pipes”):
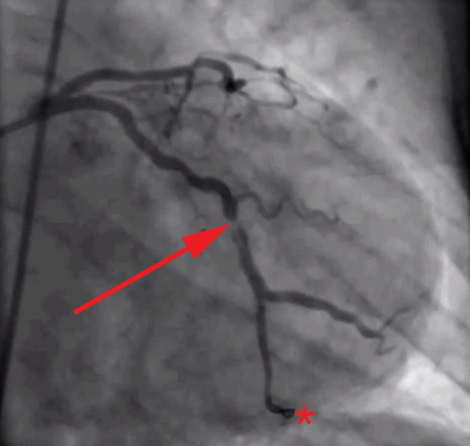
And not enough time looking at images like these (the histological, i.e., pathology, sections of coronary arteries):
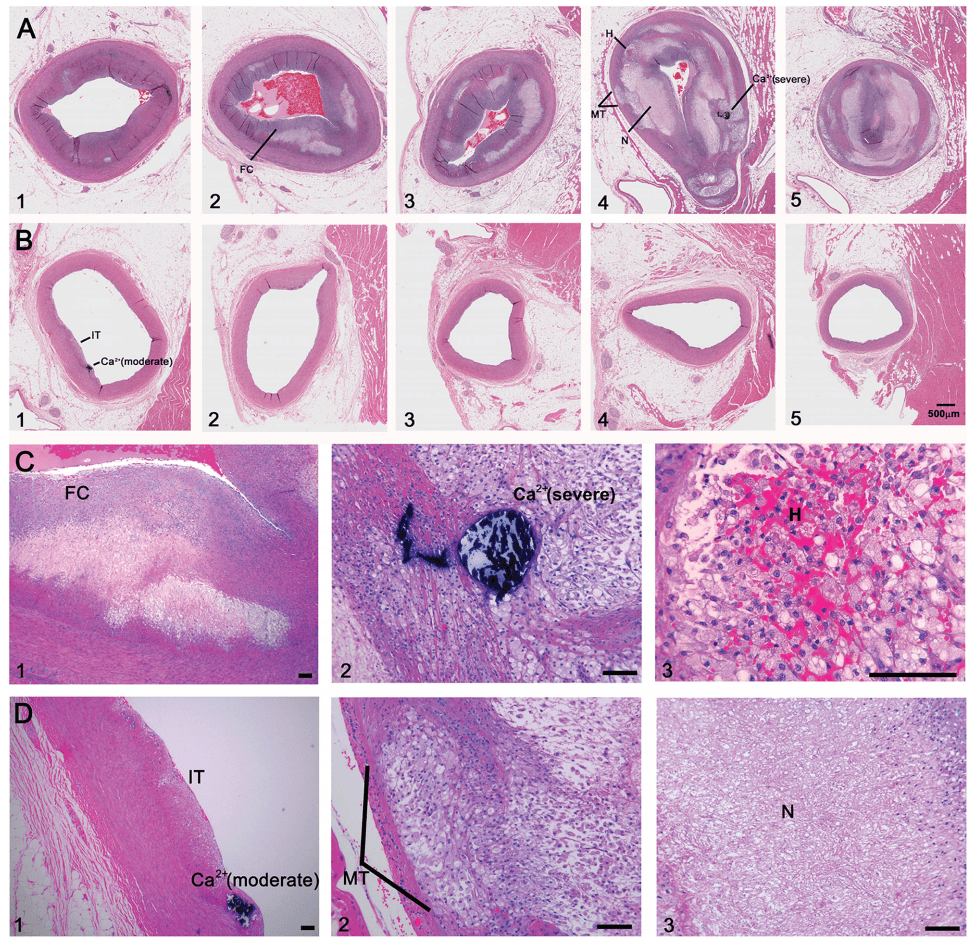
But who can blame us, I mean, angiograms are cool! But, alas, it’s time to get serious about understanding this disease if we want to prevent/delay it.
Atherosclerosis, for the cognoscenti
Ok, so now let’s get rigorous about the pathology that kills more Americans than any other disease. To understand this, as Frederic Bastiat wrote long ago, we must resort to “long and arid dissertations.” Buckle up.
The following figures were constructed from a video Tom Dayspring produced in one of his stellar lectures on the development of atherosclerosis. I’ve broken the video down into 20 or so steps which show the transition from a completely normal endothelium (i.e., at birth) through myocardial infarction. Each figure is preceded by a brief explanation of its content.
The endothelium is a protective one cell layer lining the surface of the artery lumen. Endothelial cells perform many complex functions and are capable of modulating vascular tone, as well as inflammatory and thrombotic processes. Their function depends on many circulating and local factors.

Low density lipoprotein (“LDL”) is a lipid (the bulk of which is cholesterol) transport particle. Please re-read this sentence. It is not “bad cholesterol,” a term that has no meaning. LDL—the particle—allows lipids (cholesterol, but also triglyceride, phospholipid) to be delivered through the aqueous medium of the blood, since lipids are hydrophobic (i.e., repel water) and a “carrier” is needed to transport them in blood (which is mostly water).
If LDL particles are present in physiologic (i.e., normal) concentrations, they effectively deliver cholesterol to those tissues that require it (recall: all tissues make cholesterol but some don’t make enough for their own needs and therefore cholesterol needs to be trafficked around the body).
The term LDL particle, LDL-P, and apoB are used interchangeably (the latter, because LDL particles are defined by the wrapping of a lipoprotein called apolipoprotein B-100).

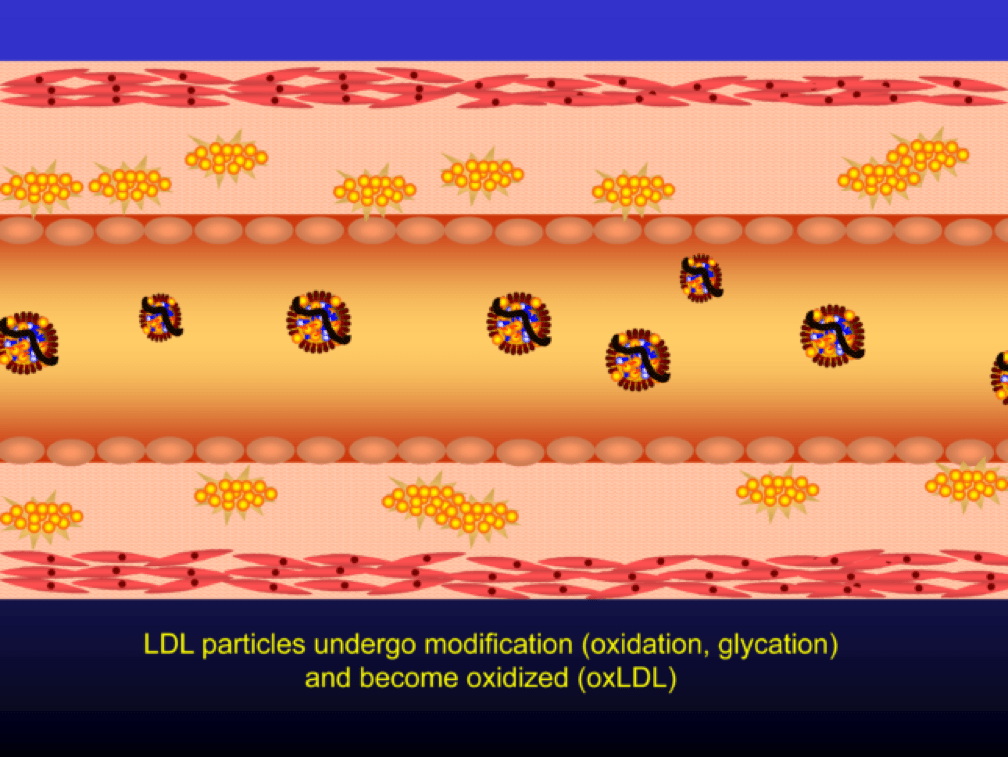
When LDL particle concentration is elevated, the lipoprotein penetrates into the subendothelial space. Once in the intimal layer, they are securely attached to intimal proteoglycan molecules. The first step in atherogenesis is surface phospholipid (PL) exposure to reactive oxygen species and oxidation of the PL. LDL particles that are not oxidized are not atherogenic. To be clear, it’s not the “getting in there” part that is the problem (HDL particles do this all the time and so do LDL particles, for that matter), it’s the “getting stuck and oxidized in there” part, formally known as retention and oxidation.
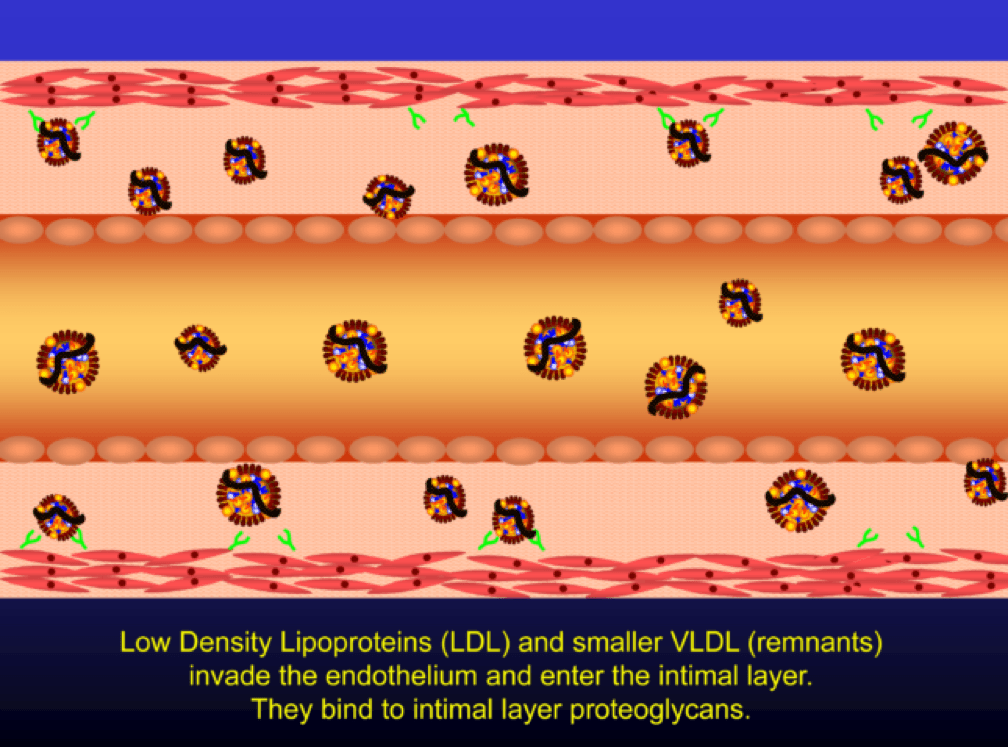
Once retained in the subendothelial space, the LDL particle may be modified, or oxidized (the clusters of yellow circles in the subendothelial space, below).
Oxidized LDL particles are toxic to the endothelium. Now-dysfunctional endothelial cells express selectins and vascular cell adhesion molecules (VCAMS) which mark the injured areas of the vascular wall. It is worth pausing here for a moment. This step is kind of the turning point in the story. It’s also a perfectly “normal” thing for the epithelium to do. They sense a problem and like any law-abiding tissue, they ask for help from law enforcement. Think of the selectins and VCAMS as 911-calls. The police, who show up shortly, are the monocytes.
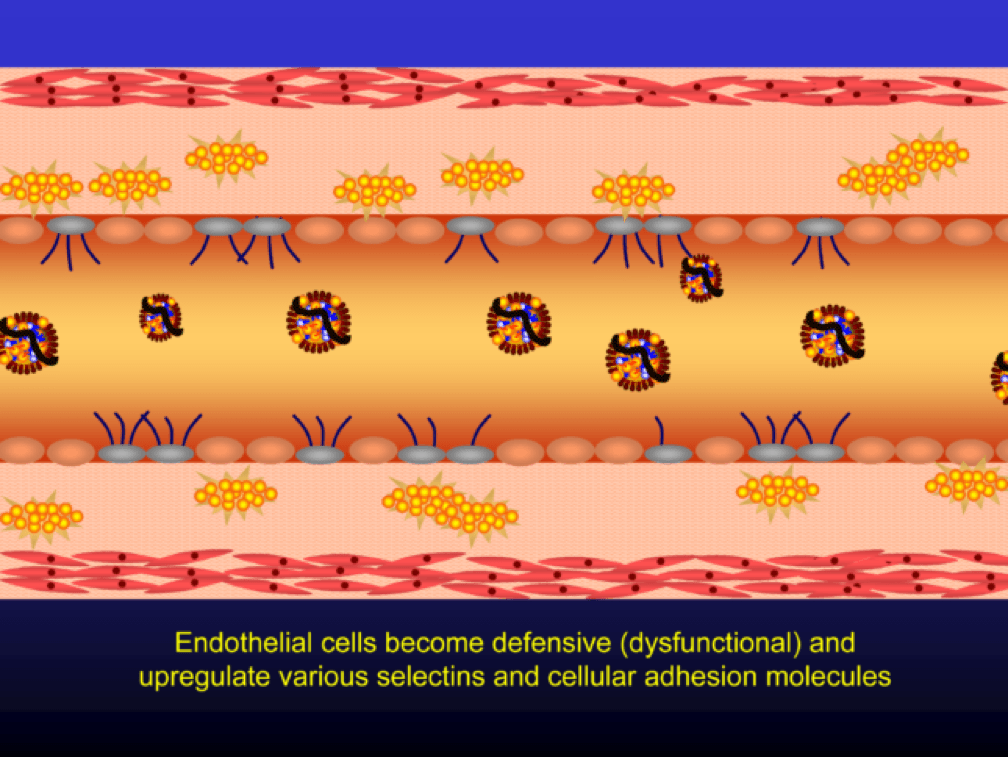
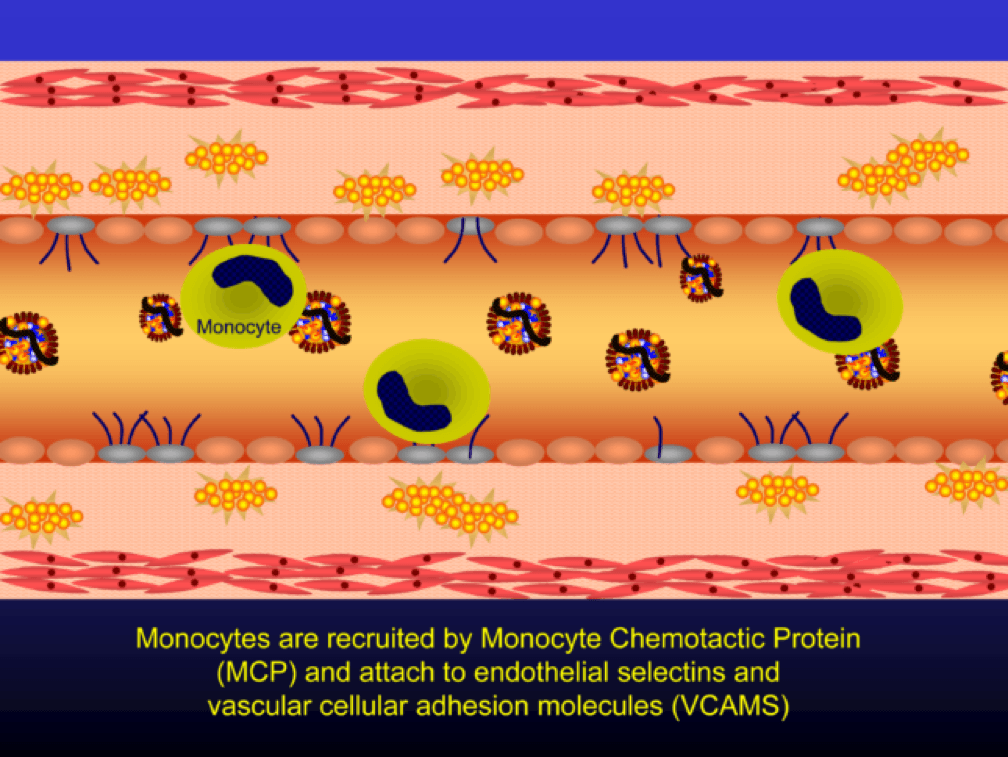
Selectins and VCAMS increase monocyte adherence to the endothelium.
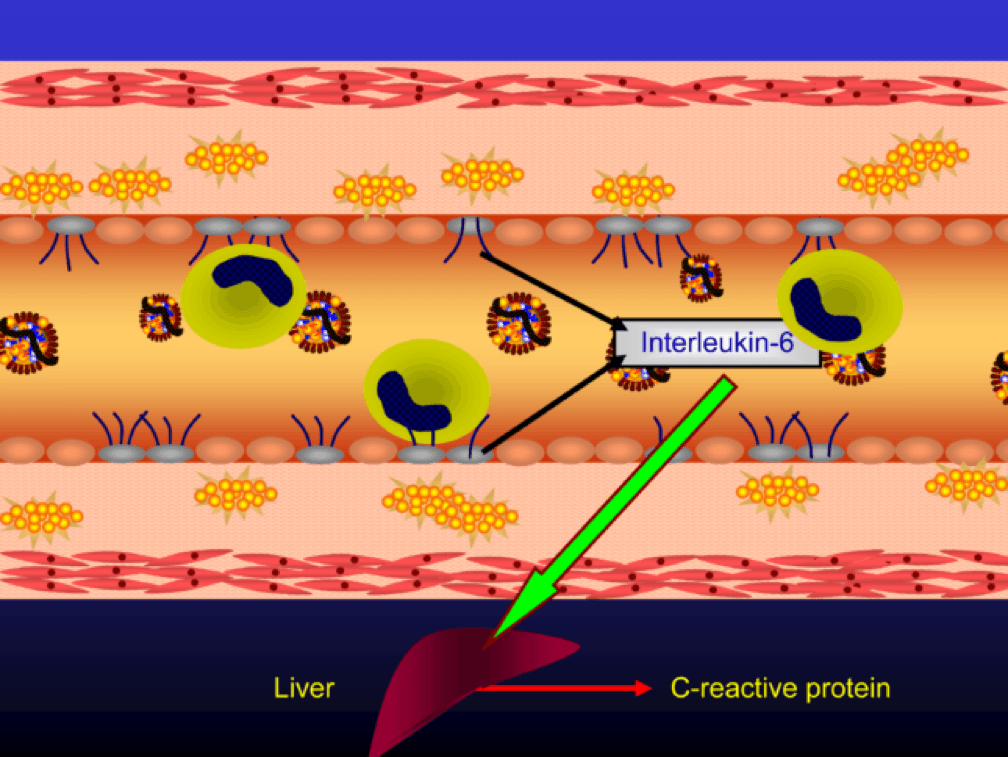
The endothelial cells also express messenger cytokines such as interlukin-6 (IL-6) and tumor necrosis factor (TNF) which circulate to the liver and induce the production of C-reactive protein (CRP).
Monocytes penetrate the subendothelial space…
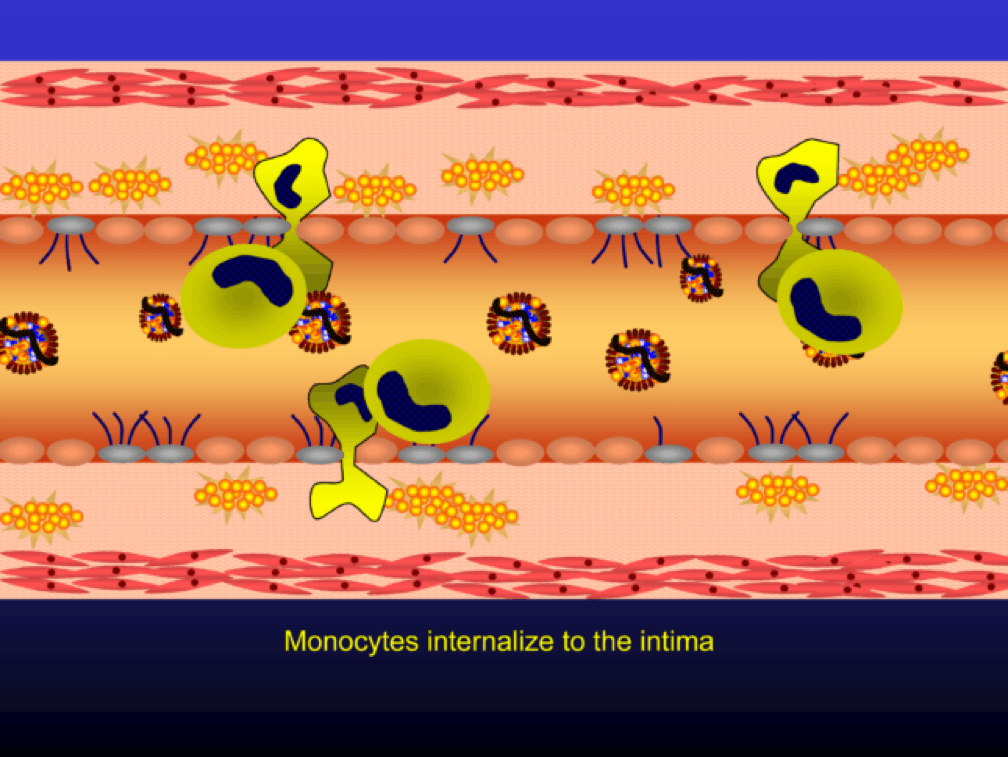
…and when they do, the monocytes differentiate into (i.e., “become”) macrophages (a more specific type of immune cell). Macrophages phagocytize (basically “ingest”) the modified or oxidized LDL particles.
The phagocytosis of oxidized LDL particles (oxLDL) and accumulation of lipid in the macrophage creates something called a foam cell.
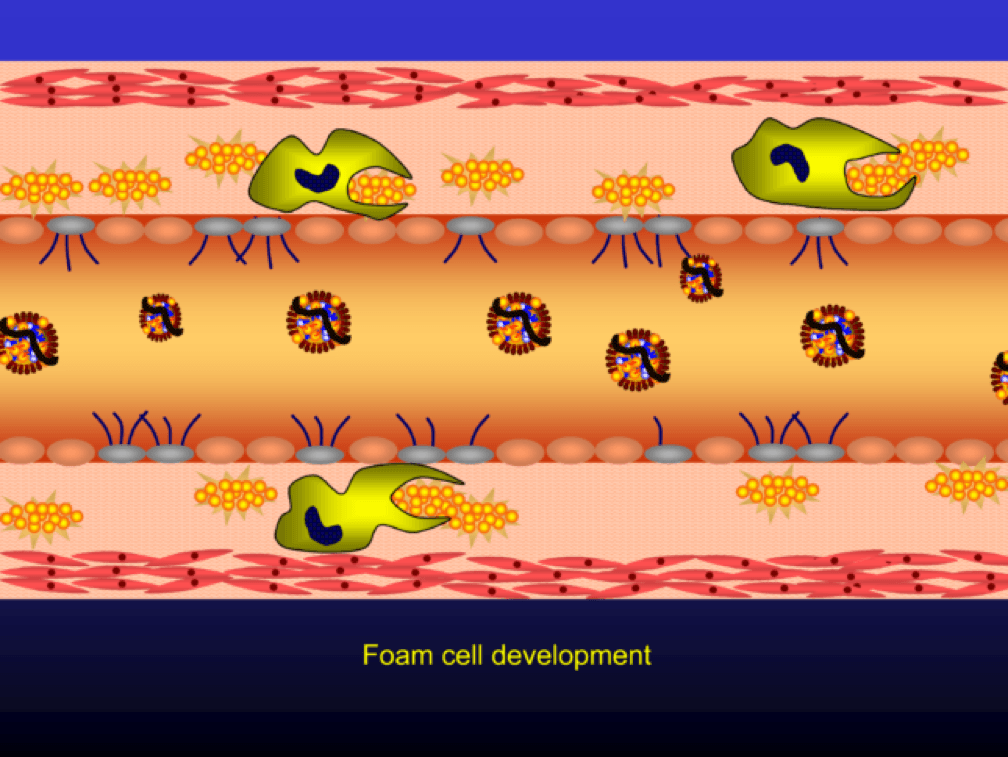
Multiple foam cells coalesce to form the characteristic fatty streak, the hallmark of an early atherosclerotic plaque. Keep this in mind. Later in this post we’ll come back to “fatty streaks” and I want you to remember how much has taken place to get us to this stage. I will posit that no one reading this post does not have fatty streaks unless there are some brilliant 5-year-olds reading this during recess..
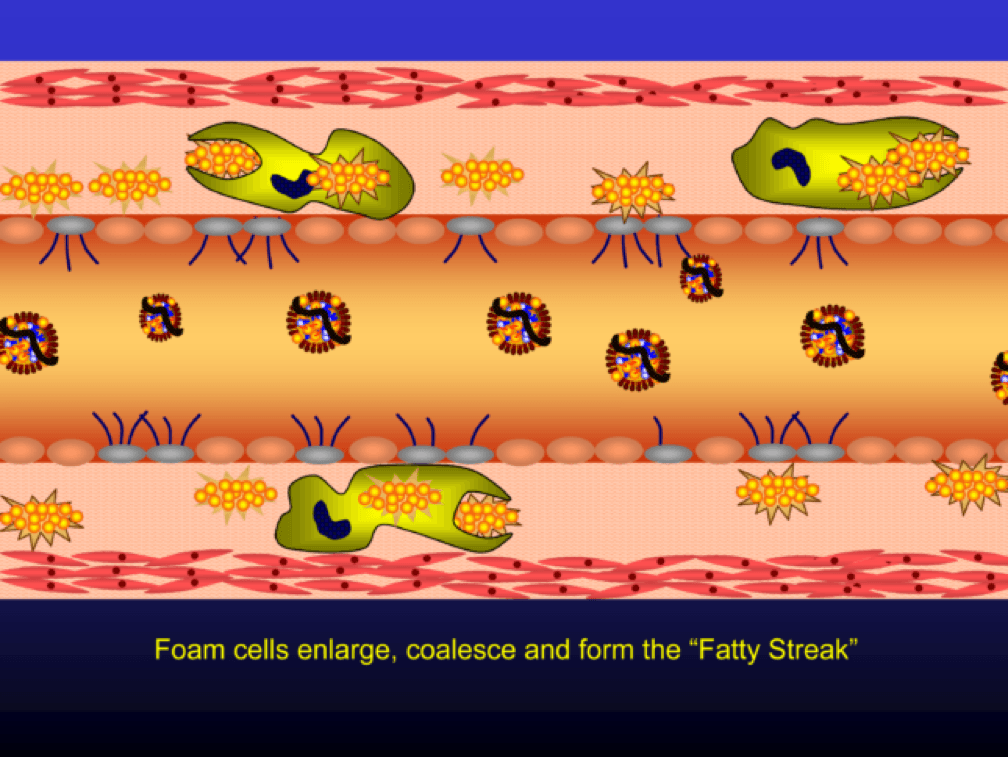
Nascent Apo A-I containing particles (also known as prebeta-HDL particles) accumulate free or unesterified cholesterol from the macrophages using ATP Binding Cassette Transporters A1.
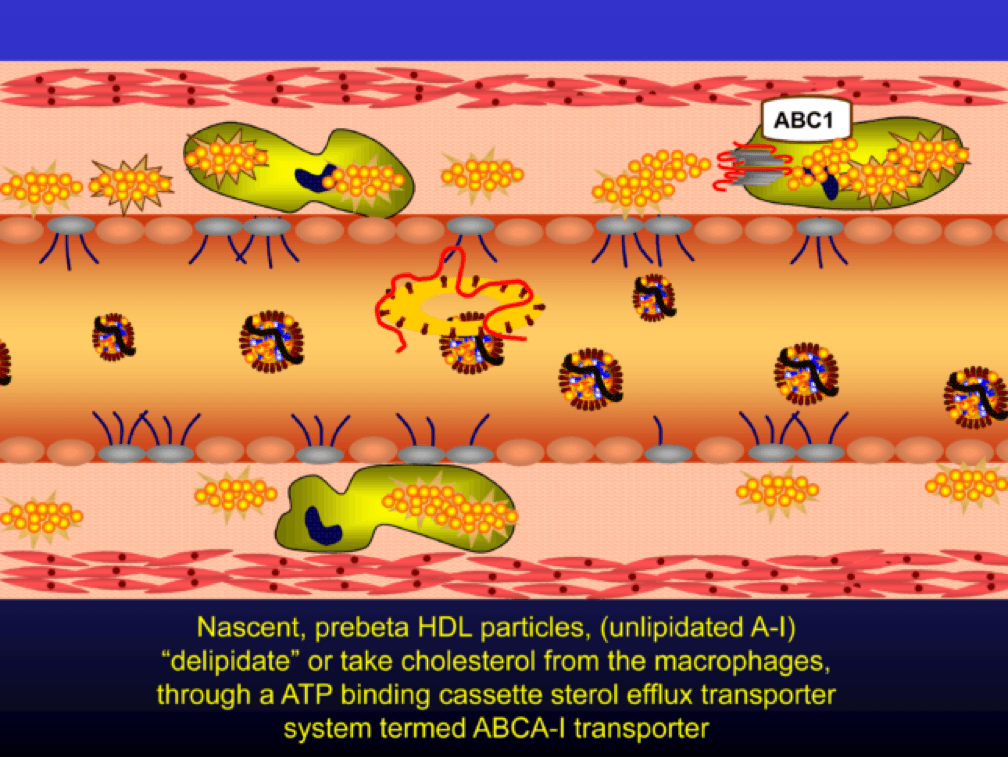
As the HDL particle lipidates itself by delipidating the macrophage (i.e., takes lipid out of the foam cell), the HDL particle, utilizing an enzyme called LCAT, esterifies the free cholesterol forming cholesteryl ester (CE). As a result, the HDL particle enlarges and is free to be delipidated at a variety of tissues. HDL delipidation occurs through multiple mechanisms using Cholesteryl Ester Transfer Protein (CETP), aqueous free diffusion, SRB1 receptors in endocrine glands or gonads (e.g., to make hormones), adipocytes (a major cholesterol storage organ) or the liver (e.g., to package for biliary delivery). HDLs can also be internalized as entire particles by surface liver receptors.
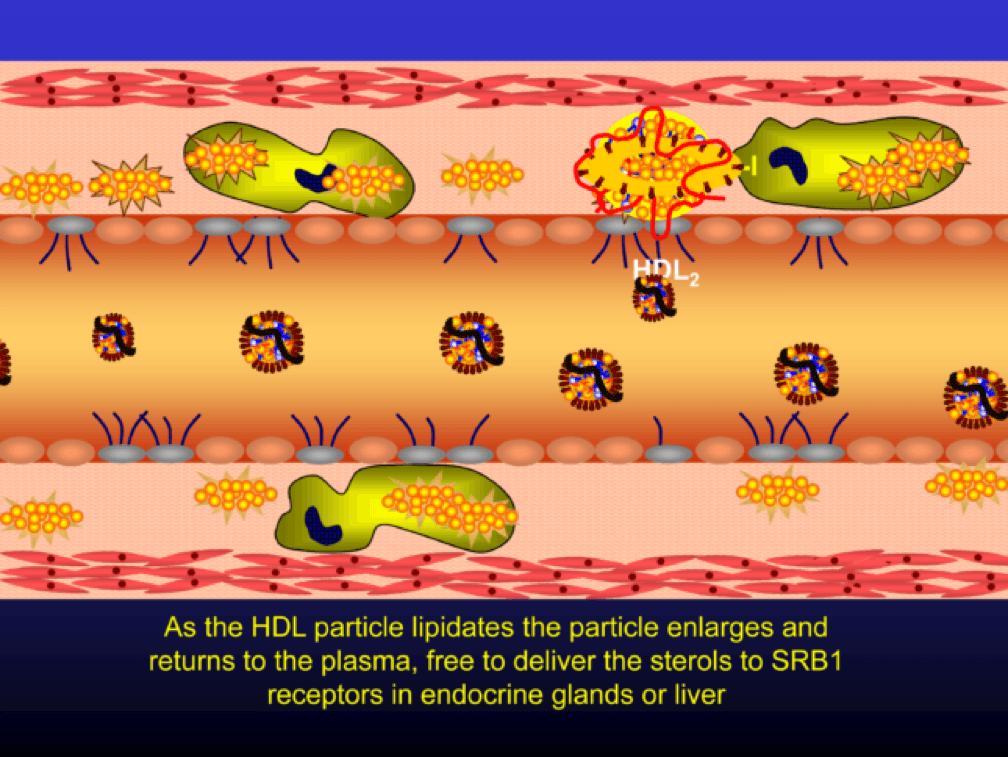
A brief, but important, digression: the complexity, above, is probably the reason why every trial that has tried to increase the concentration of cholesterol in HDL particles (i.e., raise HDL-C) has failed, and failed epically, to reduce events. The value in HDL particles (the so-called “good cholesterol”—my god I hate that term as much as I hate the term “bad cholesterol” when referring to LDL) is almost assuredly in its functional capacity—what it is doing that might be cardioprotective (very hard to measure) rather than its cholesterol content (HDL-C), which is relatively easy to measure, but probably offers zero insight other than its positive epidemiological associations. In other words, measuring HDL cholesterol content tells you little about its cholesterol efflux capacity or any other of the numerous HDL functional properties.
Macrophages that become engorged with oxLDL remain full-fledged foam cells. Foam cells produce Angiotensin II, metalloproteinases, collagenases, elastases, and other proteins, which undermine the integrity of the arterial wall, causing more endothelial dysfunction. This is where the process goes to hell. Now you’ve got a damaged barrier and the looting begins. Oh, by the way, the process to date goes unnoticed by the calcium scan and CTA.
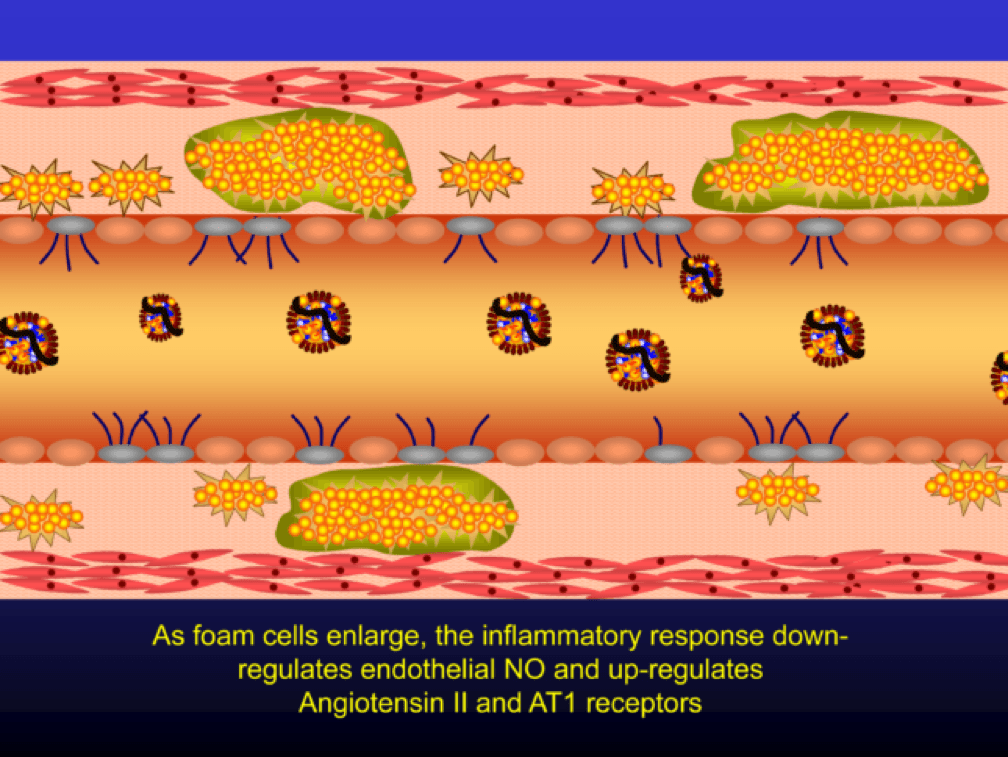
Chemotactic factors (i.e., chemical signals) trigger migration of smooth muscle cells to the area of injury in an attempt to repair the damage and pervert further disruption. Again, all of this is taking place in good faith on the part of the immune system. The smooth muscle cells are transformed into secretory cells that lay down a matrix to heal the injured wall. This matrix becomes the fibrous cap of the atherosclerotic plaque. Only now is the arterial lumen becoming encroached.

Metalloproteinases and collagenases are upregulated and they start to dissolve or weaken the plaque cap, typically at the shoulder regions where the diseased endothelial cells meet healthy endothelial cells. Some have used the term “vulnerable” to describe such plaques, which may be a correct term, but it also gives a false sense of confidence that we can treat atherosclerosis on a lesion-by-lesion basis. History has taught us that such hubris is unwarranted. Until proven otherwise, atherosclerosis should be viewed as a systemic condition of the arterial system. To see one of the best (and my favorite) papers on this topic, look no further than to this paper by Armin Arbab-Zedeh and the venerable Valentin Fuster, aptly titled “The Myth of the Vulnerable Plaque.”
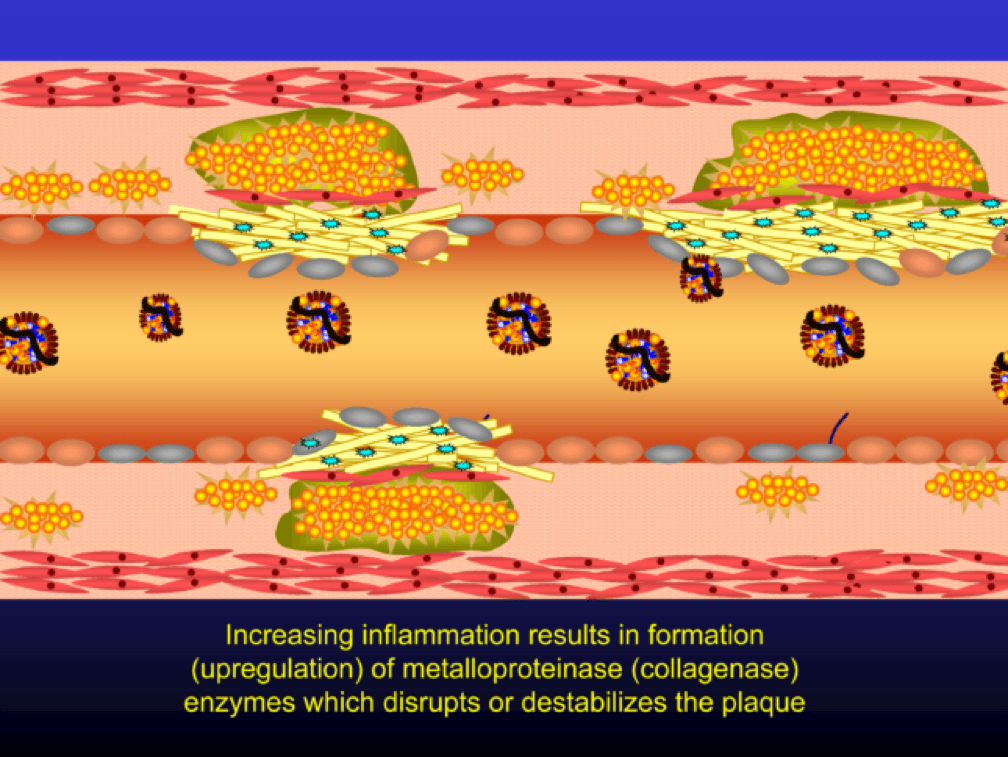
The plaque can become obstructive—i.e., it can obstruct the lumen—over time. Lipid rich plaques are unstable, and can rupture. Platelets adhere to the ruptured surface of the plaque through electrostatic factors and through binding to specific ligands.
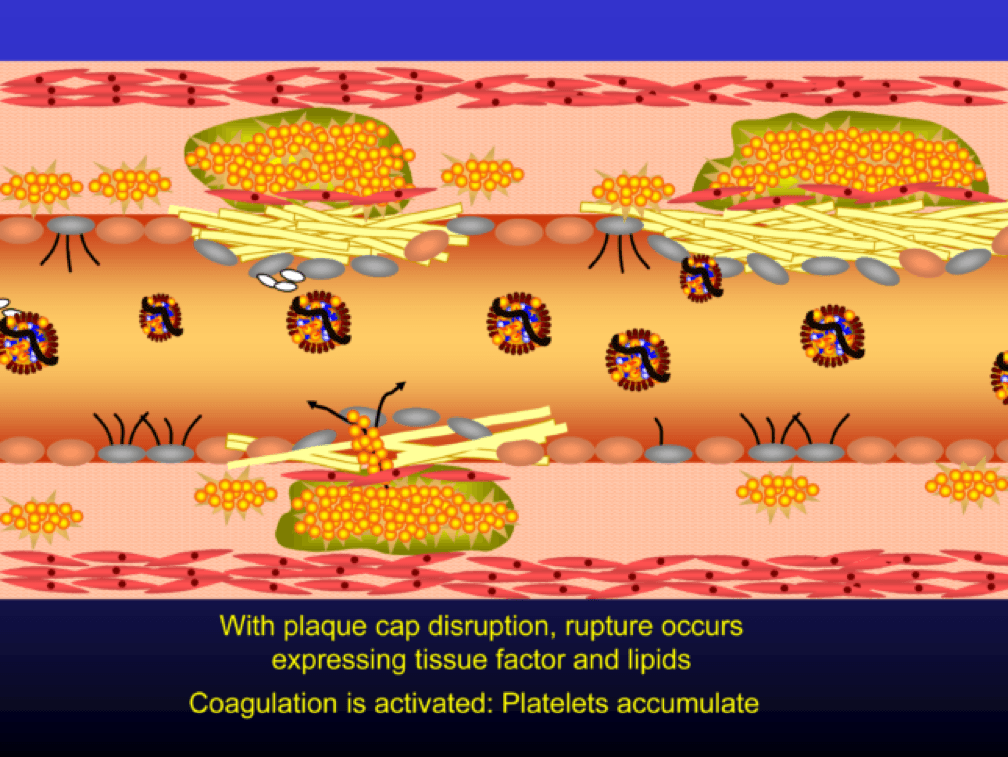
The platelets then serve as a cradle for the coagulation cascade to produce a net of fibrin (the white “net” in the figure), leading to a red clot, as red blood cells are caught in the net. A non-obstructive plaque can lead to a clinical event after the superimposition of red and white thrombus, which can occur quickly and without warning. The degree of stenosis (luminal narrowing) does not predict when this will happen.
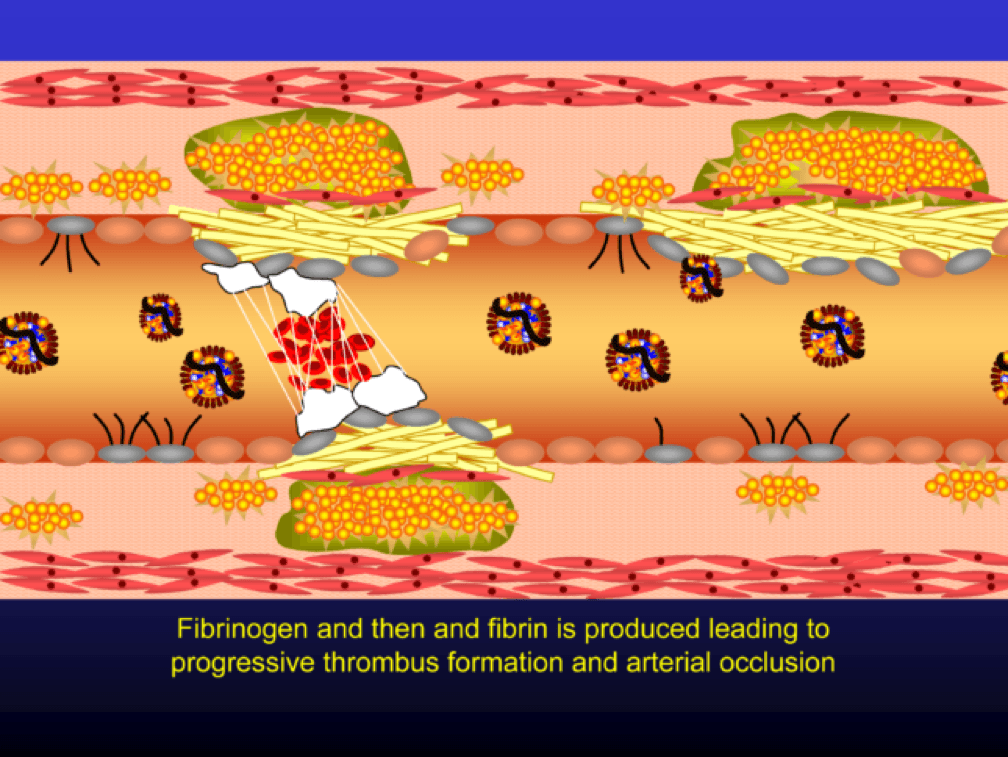
Why all of this matters
So back to this impetus for this post—Allan’s editorial. When I first met Dr. Allan Sniderman, we immediately hit it off. Like Tom, Allan has been a remarkable mentor and teacher. In fact, one of the gifts Allan gave me a while back was his personal copy of Herbert Stary’s legendary pathology textbook on atherosclerosis, Atlas of Atherosclerosis Progression and Regression. I devoured this textbook and have since purchased additional copies to always have one on hand. Ask my patients…most of them have had to sit through viewings of it like it was a vacation photo album.
Allan and I were having dinner and discussing our favorite topic. Allan asked me to guess what fraction of cardiac events (“event” is a pretty common word in the vernacular of cardiovascular disease and basically refers to a Q-wave MI, the need for re-vascularization, or cardiac death) take place in North America in those younger than 65. I knew it was a loaded question, so I rounded my guess up to 25%. I was wrong. How wrong I wouldn’t find out until Allan and his colleagues completed their analysis which formed the basis for their editorial recently published in JAMA Cardiology. Add it to your weekly reading list.
You don’t have to be a lipidologist or a pathologist to understand this paper, but it helps to understand the basics of math—big denominators can drown out even modest numerators. The aggregate figure from this paper is one of the most elegant representations of a dot product. The subfigure on the left is how most doctors (myself included until the 2nd decade of the 21st century) and authorities think of coronary heart disease (CHD)—it’s virtually silent until the 7th or 8th decade of life. What folks who think this way miss is the middle figure—the population base (the “denominator”) is shrinking while the incidence (the “numerator”) is rising. In a situation like this, the only way to really see what’s happening is to do what Sniderman et al. did—calculate the absolute event rate, as shown on the right.
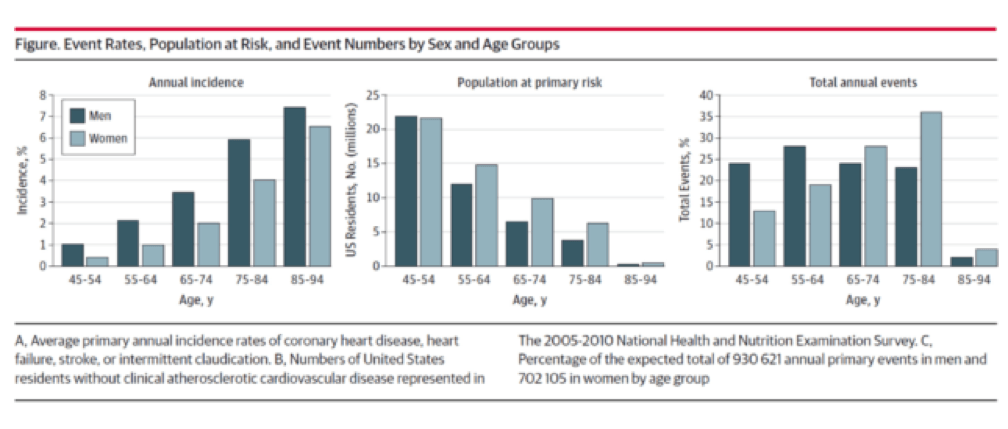
Yes, you’re reading this graph correctly. A little over half of all events in men (24% + 28%) and a little less than a third of events in women (13% + 19%) take place below the age of 65.
Tying these two insights together
Insight #1: Atherosclerosis takes a long time to evolve, and involves many steps.
Insight #2: Many cardiovascular events—half in men and one-third in women—take place in young people (i.e., those 64 or younger)
How do we reconcile these findings? Enter the pathologists. As I mentioned above, my first year pathology professor in med school insisted that pathologists were the only doctors who really understood heart disease, because they actually did the autopsies and examined the coronary arteries under microscopes.
And this brings me to my point. The only way Insight #1 and Insight #2 can be correct is if atherosclerosis takes a long time to develop. Any guesses as to what the greatest single risk factor is for heart disease? Smoking? Nope. High blood pressure? Nope. The wickedly deadly particle I have yet to write the most deserving post on, Lp(a)? Nope. LDL-P or apoB? Nope. LDL-C? I thought I told you to never say that again. CRP? Nope. None of these things. It’s age. Age trumps everything. In this sense, atherosclerosis is an “integral” disease (in the calculus sense of the word)—meaning it’s a disease of compounding injuries, as I painstakingly went through above. Age = persistent exposure to LDL-P/apoB.
Just like wealth is compounded in a highly non-linear way, so too is illness and no disease to my knowledge does so more clearly than atherosclerosis. So the jugular question is when do we need to start treating patients? That is a question I can’t answer for you. Not because I don’t have a point of view, which I most certainly do, but because it comes down to risk tolerance. I can no more impart my world view of this problem on you (though the answer seems painfully obvious to me) as I can my world view of how to raise kids or combat ISIS. But I do hope to leave you with a clear picture, at least of the disease process.
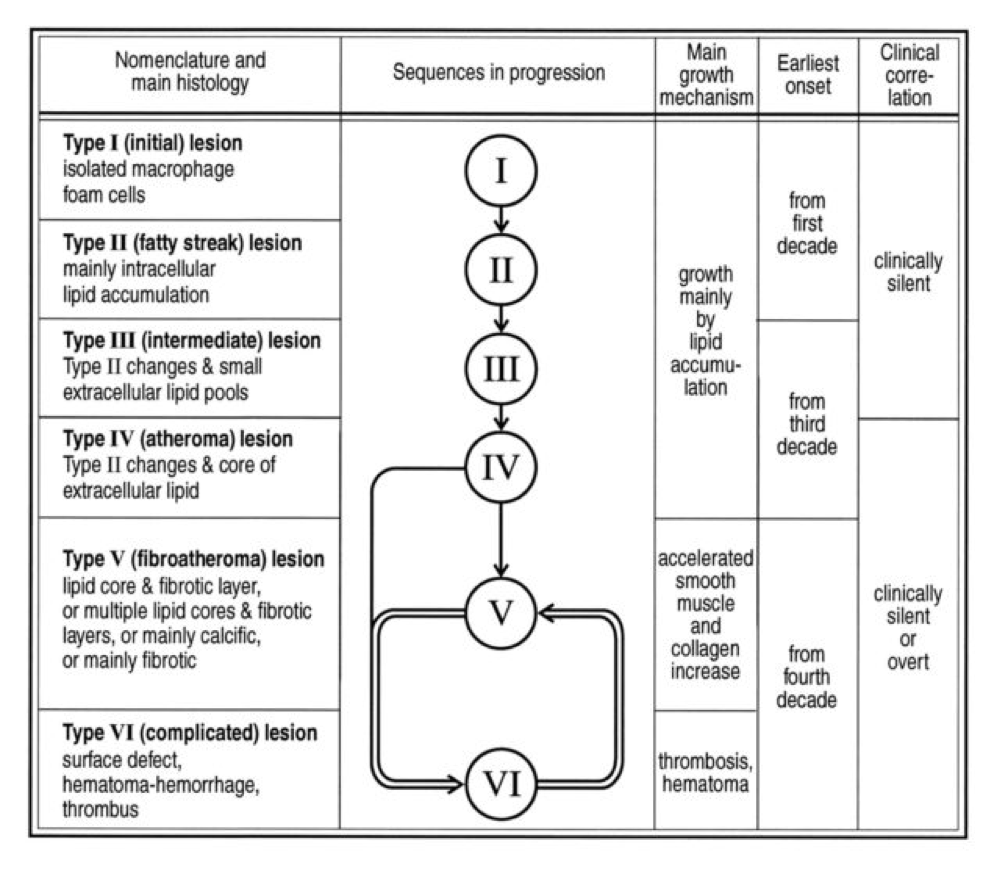
Perhaps the greatest insights into the pathogenesis of atherosclerosis, especially as they pertain to age, comes via autopsies of two variants: those of people known to die of some cause other than heart disease and those known or suspected of cardiac death. The table, below, taken from this paper, summarizes the six stages of atherosclerosis. Stary’s stages are identical, except that he further divides the sixth of these stages into three stages, for a total of eight stages, but the points remain the same. The points being, of course:
- In the first decade of life fatty streaks are being formed. That’s right, before the age of 10.
- Atheromas are present by the time most people are in their 20’s.
- By the time you are in your 30’s you are quite likely to have fibroatheroma formation.
- The vast majority of atherosclerosis-initiating sterols get into the artery as “passengers” in apoB-containing particles most of which (90%) are LDL particles.
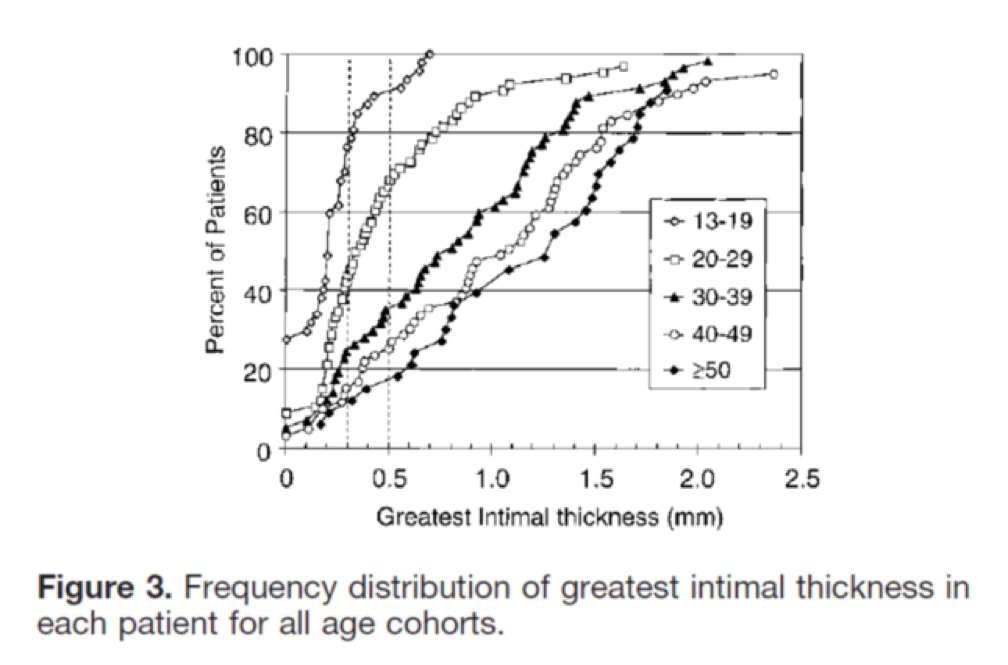
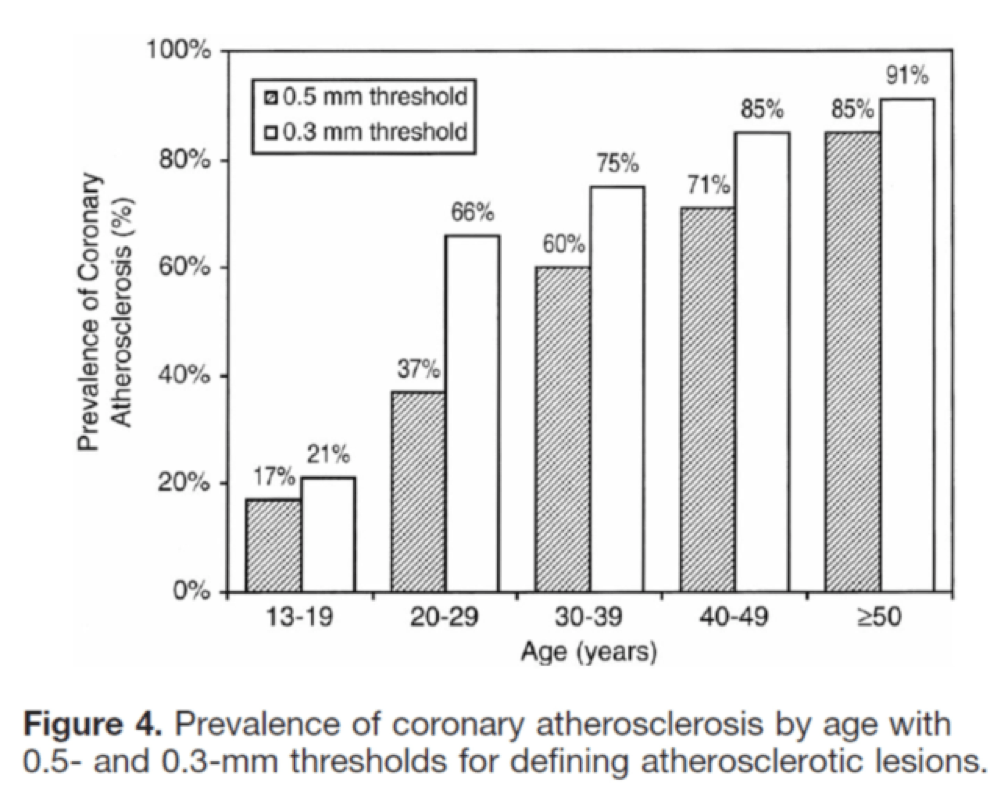
I’ll close with another interesting study, published in Circulation in 2001. The title of the study—High Prevalence of Coronary Atherosclerosis in Asymptomatic Teenagers and Young Adults—pretty much tells you what they found. The authors performed intravascular ultrasound (IVUS) on 262 heart transplant recipients about a month following their transplant. Before going any further, it’s worth pointing out that IVUS is not as sensitive as pathologic sectioning so this study is likely underestimating the degree of disease present in the hearts (but obviously one can’t section the coronary arteries of donor hearts). By doing the IVUS on the recipients so soon after they received their transplant the authors were able to study the hearts of the donors, many of whom were quite young.
The figures below, both taken from the paper, show the frequency distribution and prevalence of intimal thickening for each age cohort of donors. Consistent with the pathology studies, which actually cut open the arteries and examined them histologically, the IVUS study found a similar trend. Namely, atherosclerosis is starting much sooner than previously recognized. In this series of 262 heart donors, one out of six hearts donated by a teenager was found to have clinically measurable atherosclerosis. The authors conclude: “These findings suggest the need for intensive efforts at coronary disease prevention in young adults.”
Final thoughts
I was 35 years old, the year my daughter was born, when I first confronted my own inevitable demise which, based on my family history, was likely to be a cardiovascular one. I’m sure I’d be better off if I had that epiphany when I was 25 or possibly 15, but I’m glad it happened when it did. Today, I manage all modifiable risk factors to the level of my risk appetite and interpretation of the most nuanced scientific literature on cardiovascular disease. Does this mean I won’t succumb to heart disease? Of course not. But this is a stochastic game and the objective of the game is to increase the odds in your favor while delaying the onset of bad outcomes. It’s up to each person, and his or her doctor if necessary, to determine how aggressively he or she wants to confront the inevitable—we all have atherosclerosis at some level.
Addendum
Let me add another thought that may address some concerns.
Clearly there are factors here we (at least I) do not understand. Why do some people just stop with fatty streaks and stay that way for decades, while others progress to clinically significant heart disease? I’m sure if one normalizes for LDL-P, Lp(a)-P, and even particle size, the answer will still elude us (me). I think what people are missing here is that there is a difference between what we can infer from population-based (i.e., heterogeneous) statistics–clinical trials with drugs, epidemiology, ecology, hundreds of autopsies–and what happens at the individual level. Furthermore, the only probabilities that matter when it comes to an individual are “0” and “1”–in other words we go from the most heterogeneous to the most homogeneous.
I’m reminded of an analogy Allan Sniderman shared with me: If you’re a parent, with kids in your home, and want to know the probability that your child (or any child in your home) will die from an accidental gun shot wound, you can look at the stats, which are readily available. They will say the risk is actually pretty low–which it is on average. But what if we now adjust the information (in the Bayesian sense of the word) and note that you have a loaded .45 under your pillow (you know, for home protection). Now what happens to the risk? Of course it goes up dramatically. Why? Not just because we have “more” information–we actually have very specific “new” information. We have information about the causative agent, the gun. LDL-P and Lp(a)-P are the causative agents, and while other forces matter (e.g., inflammatory response), there is no getting around the fact that high LDL-P and Lp(a)-P are stacking the odds against you. The gun won’t pull the trigger on its own, but it’s still the causative agent. People are too quick to confuse “necessary and sufficient” with “necessary but not sufficient”–in both cases reducing the “necessary” component helps.
While I do not doubt that some people with high LDL-P and Lp(a)-P may be immune to the typical “rules” of atherosclerosis, we don’t have good enough tools to predict this, especially non-invasively. Ergo, the risk of doing something to lower these modifiable risks, even if that means taking drugs, must be weighed against the risk of doing nothing. This last point is often dismissed.
Most kids in homes with loaded guns will not die from those guns, but to ignore the loaded gun in that setting is crazy, in my opinion.
I’m going to remove the gun and put the odds back in my favor.