
When I wrote part I of this post, I naively assumed this would only be a two-part series. However, so many great questions and comments emerged from the discussion that I realize it’s worth spending much more time on this important and misunderstood topic. In terms of setting expectations, I suspect this series will require at least four parts.
So, back to the topic at hand…. (You may want to read or maybe reread part I for a biochemistry refresher before diving into part II.)
Is there a “metabolic advantage” to being in ketosis?
Few topics in the nutrition blogosphere generate so much vitriolic rhetoric as this one, and for reasons I can’t understand. I do suspect part of the issue is that folks don’t understand the actual question. I’ve used the term “metabolic advantage” because that’s so often what folks write, but I’m not sure it has a uniform meaning, which may be part of the debate. I think what folks mean when they argue about this topic is fat partitioning, but that’s my guess. To clarify the macro question, I’ve broken the question down into more well-defined chunks.
Does ketosis increase energy expenditure?
I am pretty sure when the average person argues for or against ketosis having a “metabolic advantage” what they are really arguing is whether or not, calorie-for-calorie, a person in ketosis has a higher resting energy expenditure. In other words, does a person in ketosis expend more energy than a person not in ketosis because of the caloric composition of what they consume/ingest?
Let me save you a lot of time and concern by offering you the answer: The question has not been addressed sufficiently in a properly controlled trial and, at best, we can look to lesser controlled trials and clinical observations to a make a best guess. Believe me, I’ve read every one of the studies on both sides of the argument, especially on the ‘no’ side, including this one by Barry Sears from which everyone in the ‘no’ camp likes to quote. This particular study sought to compare a non-ketogenic low carb (NLC) diet to a ketogenic low carb (KLC) diet (yes, saying ‘ketogenic’ and ‘low carb’ is a tautology in this context). Table 3 in this paper tells you all you need to know. Despite the study participants having food provided, the KLC group was not actually in ketosis as evidenced by their B-OHB levels. At 2-weeks (of a 6-week study) they were flirting with ketosis (B-OHB levels were 0.722 mM), but by the end of the study they were at 0.333 mM. While the difference between the two groups along this metric was statistically significant, it was clinically insignificant. That said, both groups did experience an increase in REE: about 86 kcal/day in the NLC group and about 139 kcal/day in the KCL group (this is calculated using the data in Table 3 and Table 2). These changes represented a significant increase from baseline but not from each other. In other words, this study only showed that reducing carbohydrate intake increased TEE but did not settle the ‘dose-response’ question.
This study by Sears et al. is a representative study and underscores the biggest problems with addressing this question:
- Dietary prescription (or adherence), and
- Ability to accurately measure differences in REE (or TEE).
Recall from a previous post, where I discuss the recent JAMA paper by David Ludwig and colleagues, I explain in detail that TEE = REE + TEF + AEE.
Measuring TEE is ideally done using doubly-labeled water or using a metabolic chamber, and the metabolic chamber is by far the more accurate way. A metabolic chamber is a room, typically about 30,000 liters in volume, with very sensitive devices to measure VO2 and VCO2 (oxygen consumed and carbon dioxide produced) to allow for what is known as indirect calorimetry. The reason this method is indirect is that it calculates energy expenditure indirectly from oxygen consumption and carbon dioxide production rather than directly via heat production. By comparison, when scientists need to calculate the energy content of food (which they do for such studies), the food is combusted in a bomb calorimeter and heat production is measured. This is referred to as direct calorimetry.
Subjects being evaluated in such studies will typically be housed in a metabolic ward (don’t confuse a metabolic ward with a metabolic chamber; the ward is simply a fancy hospital unit; the chamber is where the measurements are made) under strict supervision and every few days will spend an entire 24 hour period in one such chamber in complete isolation (so no other consumption of oxygen or production of carbon dioxide will interfere with the measurement). This is the ‘gold standard’ for measuring TEE, and shy of doing this it’s very difficult to measure differences within about 300 kcal/day.
Not surprisingly, virtually no studies use metabolic chambers and instead rely on short-term measurement of REE as a proxy. In fact, there are only about 14 metabolic chambers in the United States.
A broader question, which overlays this one, is whether any change in macronutrients impacts TEE.
Despite the limitations we allude to in the summary of this review, there is a growing body of recent literature (for example this study, this study, and this study) that do suggest a thermogenic effect, specifically, of a ketogenic diet, possibly through fibroblast growth factor-21 (FGF21) which increases with B-OHB production by the liver.
These mice studies (of course, what is true in mice isn’t necessarily true in humans, but it’s much easier to measure in mice) show that FGF21 expression in the liver is under the control of the transcription factor peroxisome proliferator-activated receptor a (PPARa), which is activated during starvation. Increased FGF21 promotes lipolysis in adipose tissue and the release of fatty acids into the circulation. Fatty acids are then taken up by the liver and converted into ketone bodies. FGF21 expression in liver and adipose tissue is increased not only by fasting but also by a high fat diet as well as in genetic obesity which, according to these studies, may indicate that increased FGF21 expression may be protective. Hence, ketosis may increase TEE either by increasing REE (thermogenic) or AEE (the ketogenic mice move more). Of course, this does not say why. Is the ketogenic diet, by maximally reducing insulin levels, maximally increasing lipolysis (which dissipates energy via thermogenic and/or activity ‘sinks’) or is the ketogenic diet via some other mechanism increasing thermogenesis and activity, and the increased lipolysis is simply the result? We don’t actually know yet.
Bottom line: There is sufficient clinical evidence to suggest that carbohydrate restriction may increase TEE in subjects, though there is great variability across studies (likely due the morass of poorly designed and executed studies which dilute the pool of studies coupled with the technical difficulties in measuring such changes) andwithin subjects (look at the energy expenditure charts in this post). The bigger question is if ketosis does so to a greater extent than would be expected/predicted based on just the further reduction in carbohydrate content. In other words, is there something “special” about ketosis that increases TEE beyond the dose effect of carbohydrate removal? That study has not been done properly, yet. However, I have it on very good authority that such a study is in the works, and we should have an answer in a few years (yes, it takes that long to do these studies properly).
Does ketosis offer a physical performance advantage?
Like the previous question this one needs to be defined correctly if we’re going to have any chance at addressing it. Many frameworks exist to define physical performance which center around speed, strength, agility, and endurance. For clarity, let’s consider the following metrics which are easy to define and measure
- Aerobic capacity
- Anaerobic power
- Muscular strength
- Muscular endurance
There are certainly other metrics against which to evaluate physical performance (e.g., flexibility, coordination, speed), but I haven’t seen much debate around these metrics.
To cut to the chase, the answers to these questions are probably as follows:
- Does ketosis enhance aerobic capacity? Likely
- Does ketosis enhance anaerobic power? No
- Does ketosis enhance muscular strength? Unlikely
- Does ketosis enhance muscular endurance? Likely
Why? Like the previous question about energy expenditure, addressing this question requires defining it correctly. The cleanest way to define this question, in my mind, is through the lens of substrate use, oxygen consumption, and mechanical work.
But this is tough to do! In fact, to do so cleanly requires a model where the relationship between these variables is clearly defined. Fortunately, one such model does exist: animal hearts. (Human hearts would work too, but we’re not about to subject humans to these experiments.) Several studies, such as this, this, and this, have described these techniques in all of their glorious complexities. To fully explain the mathematics is beyond the scope of this post, and not really necessary to understand the point. To illustrate this body of literature, I’ll use this article by Yashihiro Kashiwaya et al.
The heart is studied because the work action is (relatively) simple to measure: cardiac output, which is the product of stroke volume (how much blood the heart pumps out per beat) and heart rate (how many times the heart beats per minute). One can also measure oxygen consumption, all intermediate metabolites, and then calculate cardiac efficiency. Efficiency increases as work increases relative to oxygen consumption.
Before we jump into the data, you’ll need to recall two important pieces of physiology to “get” this concept: the acute (vs. chronic) metabolic effect of insulin, and the way ketone bodies enter the Krebs Cycle.
The acute metabolic effects of insulin are as follows:
- Insulin promotes translocation (movement from inside the cell to the cell membrane) of GLUT4 transporters, which facilitate the flux of glucose from the plasma into the inside of the cell.
- Insulin drives the accumulation of glycogen in muscle and liver cells, when there is capacity to do so.
- Least known by most, insulin stimulates the activity of pyruvate dehydrogenase (PDH) inside the mitochondria, thereby increasing the conversion of pyruvate to acetyl CoA (see figure below).
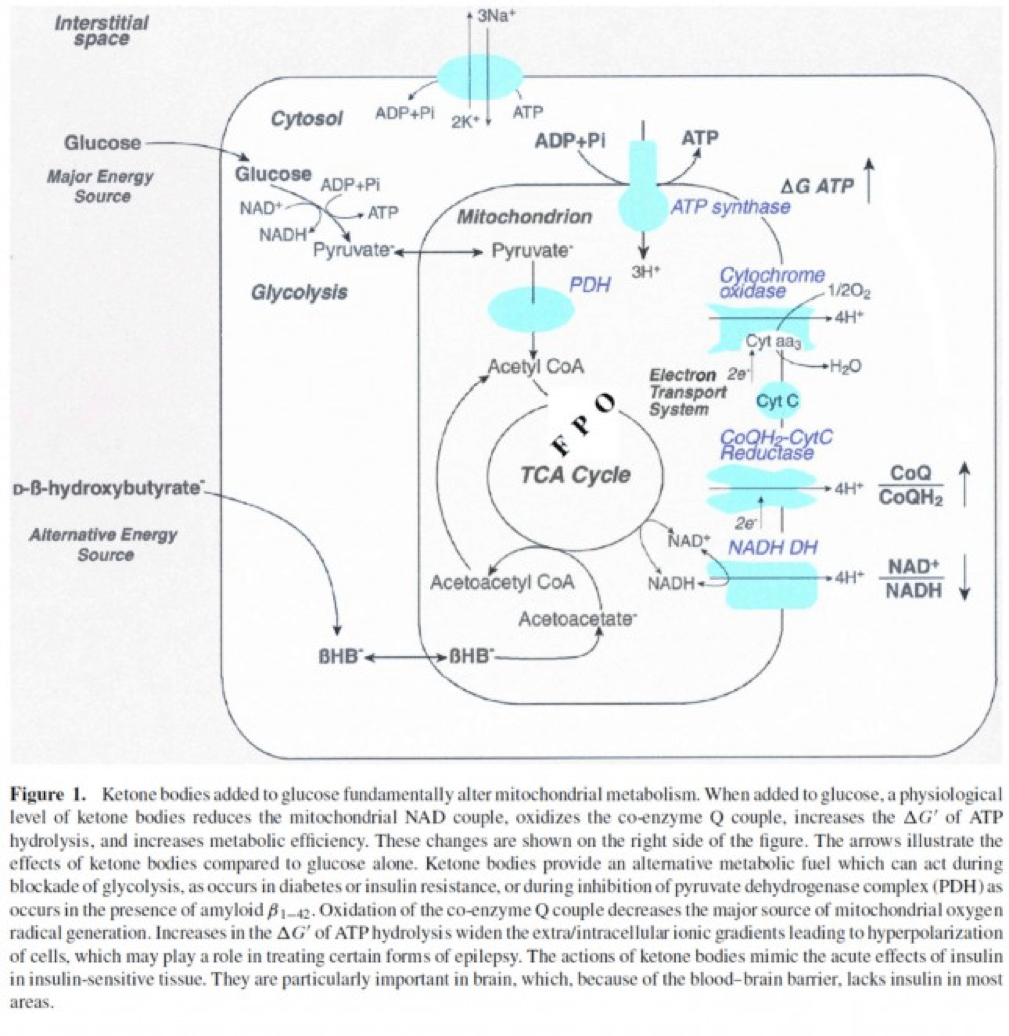
The second important point to recall is that ketone bodies bypass this process (i.e., glucose to pyruvate to acetyl CoA), as B-OHB enters the mitochondria, converts into acetoacetate, and enters the Krebs Cycle directly (between succinyl CoA and succinate, for any biochem wonks out there). I keep alluding to this distinction for a reason that will become clear shortly.
An elegant way to test the relative impact of glucose, insulin, and B-OHB on muscular efficiency is to “treat” a perfused rat heart under the following four conditions:
- Glucose alone (G)
- Glucose + insulin (GI)
- Glucose + B-OHB (GK)
- Glucose + insulin + B-OHB (GIK)
In fact, that’s exactly what this paper did. Look at what they found:
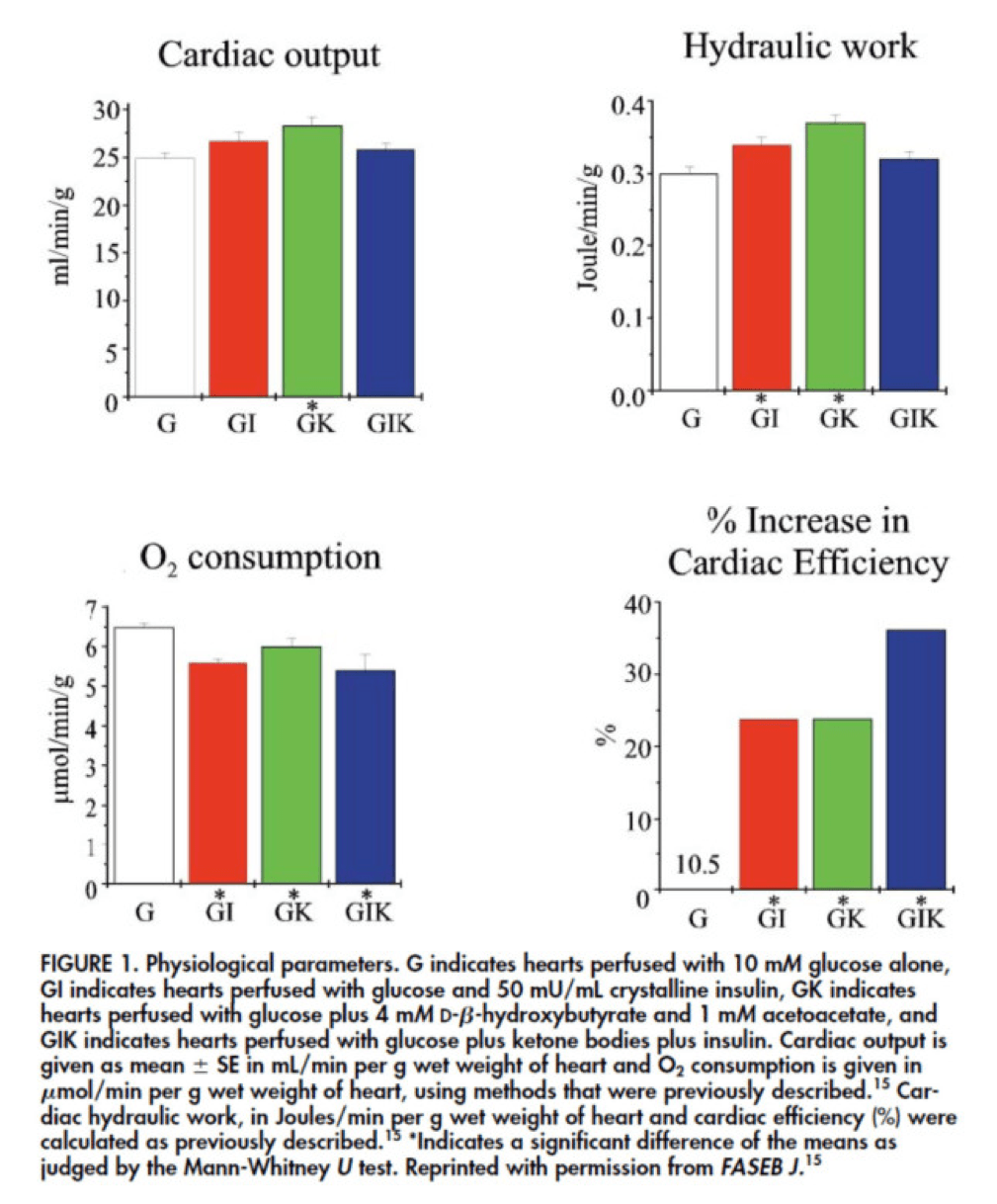
The upper two graphs in this figure show similar information, namely the response of cardiac output and hydraulic work to each treatment. (Cardiac output is pure measurement, as I described above, of volume of blood displaced per unit time. Hydraulic work is a bit more nuanced; it measures the mechanical work being done by the fluid.)
Adding insulin to a fixed glucose (GI) load increases both cardiac output and hydraulic work, but it’s only significant in the case of hydraulic work. Conversely, adding B-OHB to glucose (GK) increases both cardiac output and hydraulic work significantly. Interestingly, combining insulin and B-OHB with glucose (GIK) increases neither.
Oxygen consumption was significantly reduced in all arms relative to glucose alone, so we expect the cardiac efficiency to be much higher in all states. (Why? Because for less oxygen consumption, the hearts were able to deliver greater cardiac output and accomplish greater hydraulic work.)
The figure on the bottom right shows this exactly. If you’re wondering why the gain in efficiency is so great (24-37%), the answer is not evident from this figure. To understand exactly how and why adding high amounts of insulin (50 uU/mL) or B-OHB (4 mM) to glucose (10 mM) could cause such a step-function increase in cardiac efficiency, you need to look specifically at how the concentration of metabolic intermediates (e.g., ATP, ADP, lactate) varied in the rat heart cells.
This is where this post goes from “kind of technical” to “really technical.”
The figure below presents the results from this analysis. The height of the bar shows the fold-increase for each of the three treatments relative to glucose alone. To orient you, let’s look at a few examples. In the upper left of the figure you’ll note that GI and GIK both significantly increase glucose concentration in the cell, while GK does not. Why? The GI and GIK treatments both increase the number of GLUT4 transporters translocated to the cell surface so more glucose can flux in. GK does increase glucose concentration, but not significantly (in the statistical sense).
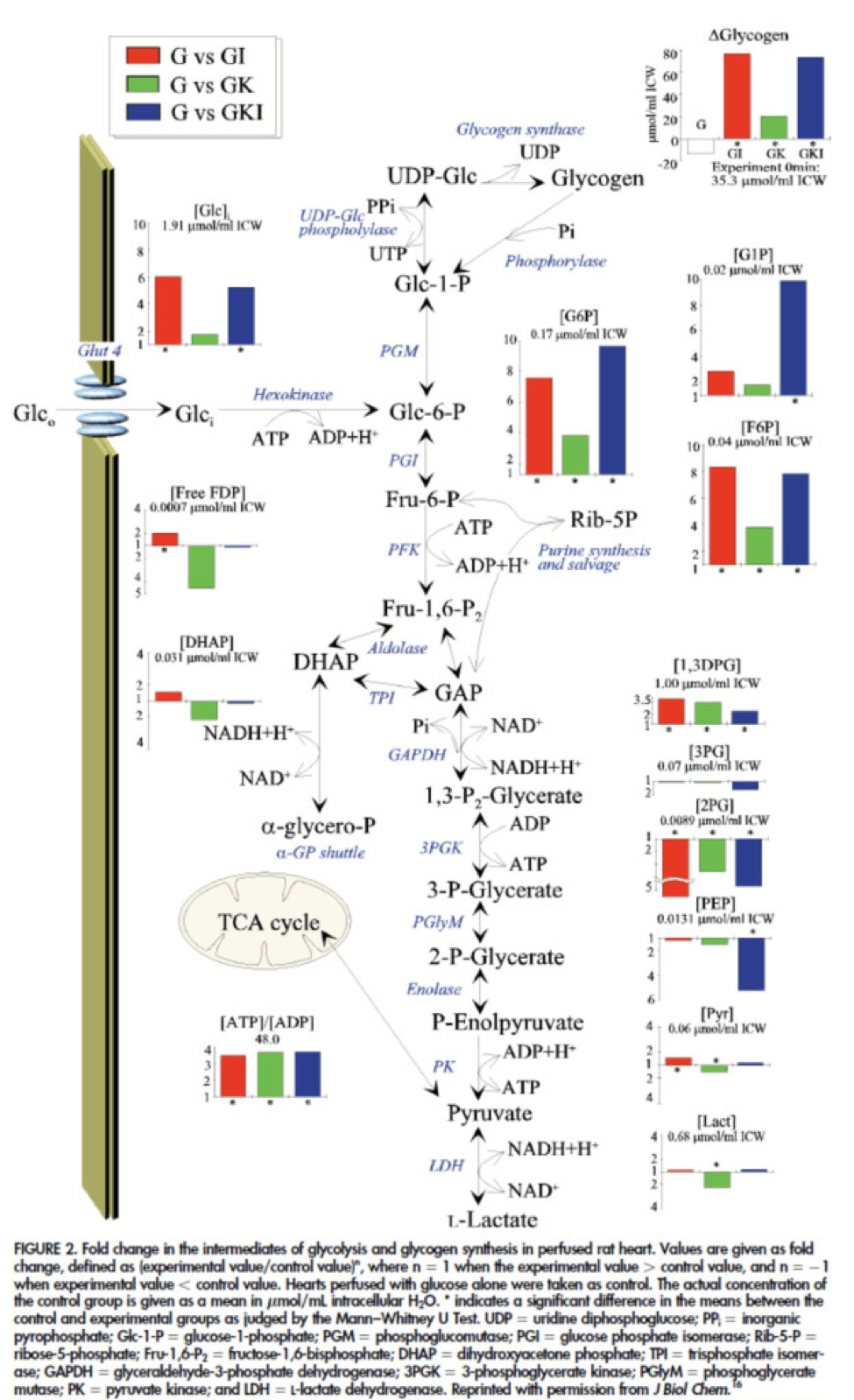
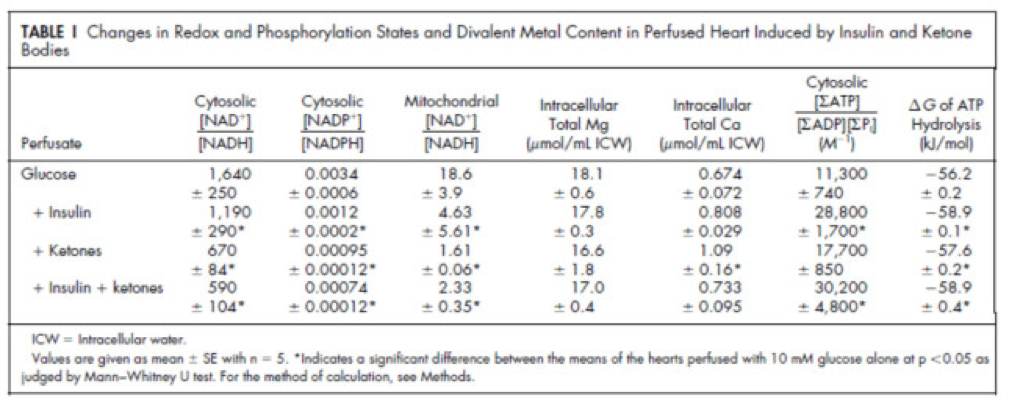
Table 1 from this paper, below, summarizes the important changes from this analysis. In particular, look at the last column, the Delta G of ATP hydrolysis.
I was really hoping to write this post without ever having to explain Delta G, but alas, I’ve decided to do it for two reasons:
- To really “get” this concept, we can’t avoid it, and;
- The readers of this blog are smart enough to handle this concept.
Delta G, or Gibbs free energy, is the “free” (though a better term is probably “available” or “potential”) energy of a system.
Delta G = Delta H – Temperature * Delta S, where H is enthalpy and S is entropy. The more negative Delta G is, the more available (or potential or “free”) energy exists in the system (e.g., a Delta G of -1000 kcal/mol has more available energy than one of -500 kcal/mol). To help with the point I really want to make I refer to you this video which does a good job explaining Gibbs free energy in the context of a biologic system. Take a moment to watch this video, if you’re not already intimately familiar with this concept.
Now that you understand Delta G, you will appreciate the significance of the table above. The Gibbs free energy of the GI, GK, and GIK states are all more negative than that of just glucose. In other words, these interventions offer more potential energy (with less oxygen consumption, don’t forget, which is the really amazing part).
To see what the substrate-by-substrate changes look like across the mitochondria and ETC, look at this figure:
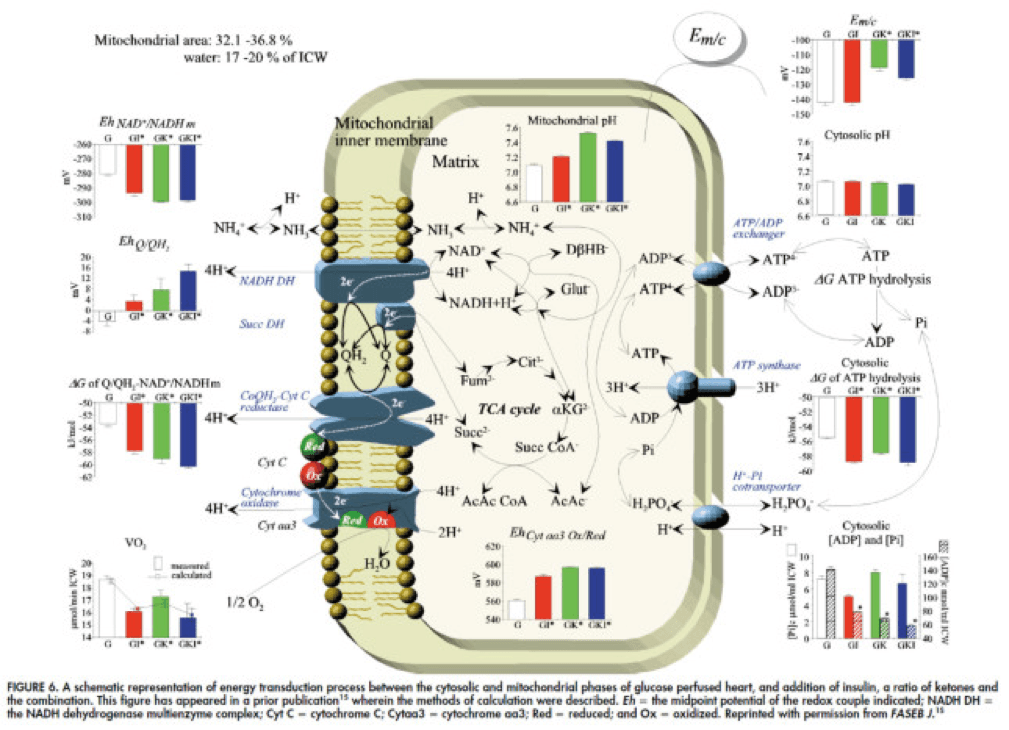
Though it is by no means remotely obvious, what is happening above boils down to two major shifts in substrate utilization:
- In one step the reactants NADH/NAD+ become more reduced (in the chemical sense), and;
- In another step the reactants CoQ/CoQH2 become more oxidized (in the chemical sense).
These changes, taken together, widen the energetic gap between the states and, in turn, translates to a higher (i.e., more negative) Delta G which translates to greater ATP production per unit of carbon.
Additional work, which you’ll be delighted to know I will not detail here, in fact shows that on a per carbon basis, B-OHB generates more ATP per 2-carbon moiety than glucose or pyruvate. As an aside, this phenomenon was first described in 1945 by the late Henry Lardy, who observed that sperm motility increased in the presence of B-OHB (relative to glucose) while oxygen consumption decreased!
Is there a reason to prefer GK over GI?
Yes. Recall that ketones make their way onto the metabolic playing field without going through PDH. Adding more insulin to the equation forces more pyruvate towards PDH into acetyl CoA. While B-OHB “mimics” the effect of additional insulin, it does so in a much cleaner fashion without the complex cascade of events brought on by additional insulin (e.g., decreased lipolysis) and, perhaps most importantly, avoids the logjam of impaired PDH due to insulin resistance (I’ll come back to this point in a future post when I address Alzheimer’s disease and Parkinson’s disease). In essence, B-OHB “hijacks” the Krebs Cycle via a slick trick that lets it bypass the bottleneck, PDH. All the glucose and insulin in the world can’t overcome this bottleneck. It’s truly a privileged state and a remarkable evolutionary trick that we can utilize B-OHB.
Back to the original question…
Clearly, in the highly controlled setting of a perfused rat heart, ketones offer an enormous thermodynamic advantage (28%!). But what about in aggregate human performance? There is no reason to believe that therapeutic levels of B-OHB (either through nutritional ketosis or by ingesting ketone esters) would increase anaerobic power, since the anaerobic system does not leverage the Delta G improvement I’ve outlined here. Same is true for muscular strength. However, there is reason to believe that aerobic capacity and muscular endurance could be improved with sufficient B-OHB present to compliment glucose.
It turns out this has been demonstrated repeatedly in subjects ingesting ketone esters, developed by Dr. Richard Veech (NIH) and Dr. Kieran Clarke (Oxford). Because the results of their work have not yet been published, I can’t comment much or share the data I have, which they shared with me. I can say the ingestion of B-OHB in the D-isoform (the physiologic isoform), resulting in serum levels between 4 and 6 mM, did lead to significant increases in aerobic power and efficiency in several groups of elite athletes (e.g., Olympians) across multiple physical tasks maximally stressing the aerobic system.
Once published, I believe these studies will be a real shot across the bow of how we view athletic performance. It is very important to point out, however, that these studies don’t exactly address the most relevant question, which has to do with nutritional ketosis. In other words, ingesting ketone esters to a level of 4 to 6 mM might not be the same as de novo producing B-OHB to those levels. But, such trials should be forthcoming in the next few years. Personally, I am most eager to see the results of a ketone ester alone versus nutritional ketosis versus combination treatment, all to the same serum level of B-OHB.
The Hall Paradox
For the really astute readers, you may be saying, “Waaaaaaaait a minute, Peter…if ketones increase Gibbs free energy while reducing oxygen consumption, should this imply TEE goes down?” You’re right to ask this question. It was the first question I asked when I fully digested this material. If each molecule of B-OHB gives your muscles more ATP for less oxygen, you should expend less not more energy at the same caloric intake, right?
I was discussing this with Kevin Hall at NIH, an expert in metabolism and endocrinology. Kevin pointed out the error in my logic. I failed (in my question) to account for the energetic cost of making the ketones out of fat. Remember, in the experiments described above, the B-OHB is being provided for “free.” But physiologically (i.e., in nutritional ketosis or even starvation), we have to make the B-OHB out of fat. The net energy cost of doing this is actually great. According to Kevin, it is not generally appreciated how making ketones from fatty acids affects overall energy efficiency. Nevertheless, this can be examined by comparing the enthalpy of combustion of 4.5 moles of B-OHB, which is about -2,192 kcal, with the enthalpy of combustion of 1 mole of stearic acid (about -2,710 kcal) that was used to produce the 4.5 moles of ketones. Thus, there is about 20% energy loss in this process. Hence, the energy gain provided by the ketones is actually less than the energy cost of making them, at least in theory.
This suggests that being in nutritional ketosis may require more overall system energy, while still increasing work potential. In other words, a person in nutritional ketosis may increase their overall energy expenditure, while at the same time increasing their muscular efficiency. In honor of Kevin, I refer to this as the Hall Paradox.
Parting shot
Ok, if you’re still reading this, give yourself a pat on the back. This was a bit of chemistry tour de force. Why did I do it? Well, frankly, I’m tired of reading so much nonsense on this topic. Everybody with a WordPress account (and countless people without) feels entitled to spew their opinions about ketosis without even the slightest clue of what they are talking about. As I said in part I of this series, there is no bumper sticker way to address this question, so to say ketosis is “good” or “bad” without getting into the details is as useful as a warm bucket of hamster vomit (unless you’re Daniel Tosh, in which case I bet you can find a great use for it).
Next time, I’ll try to back it out of the weeds and get to more clinically interesting stuff. But we had to do this and we’re better for it.



