
Please excuse me for not falling out of my chair after reading the following headline in the New York Times on January 16th:
“High-Fat Diet May Fuel Spread of Prostate Cancer.”
Look, I get it. I remember back in the day (uh oh, here comes old-man-Attia) when I’d flip on the television in the morning as the local station was going to commercial: “A common item in your refrigerator that may be killing you. News at 5.” I remember thinking, why isn’t this breaking news? Seems pretty damn important (as I glance over at the rectangular grim reaper, wondering what’s the culprit in there).
We start to get that the news is not just the news. There’s a business behind the news that wants to get our attention and they’re not above sensationalism in order to get it. At some point I think a lot of us sort of tune out to this stuff. As someone who practices medicine and researches health and disease, I don’t afford myself that luxury.
Technically, the title in the Times article is not a false statement. (Adding the modal verb “may” is a nice way of hedging your bets.) Do you think the Times (or I) would title a piece “An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer?” Not likely. We leave that up to Nature Genetics.
I thought it might be useful to share a little running diary by reading and commenting on some of the passages by Gina Kolata, author of the Times article, and see if it holds up to scrutiny.1This requires reading the source article in Nature Genetics. I recommend you do this in general, but this one in particular. It has a lot of moving parts, a number of studies within the studies, so to speak. Assays and analyses galore. I’ll frame this post with some questions that can be applied to similar news stories on health and disease.
The word semantics often gets thrown around as a pejorative in everyday life. However, meaning and context are the sine qua non of good science, and arguably good science reporting as well. So, let’s quibble over semantics, shall we?
The Times article begins:
Obesity is linked to prostate cancer, scientists know, but it’s not clear why. On Monday, researchers reported a surprising connection.
When prostate cancers lose a particular gene, they become tiny fat factories, a team at Beth Israel Deaconess Medical Center in Boston reported in a paper published in Nature Genetics.
Then the cancers spread from the prostate, often with deadly effect. Prostate cancers that have not lost that gene also can spread, or metastasize — in mice, at least — but only if they have a ready source of fat from the diet.
That finding suggests that dietary fat can substitute for the loss of the gene, fueling prostate cancer. Moreover, the investigators found, an obesity drug that blocks fat production can make metastatic prostate cancers regress in mice and prevent them from spreading.
Actually, there is disagreement among researchers on whether there’s a link between obesity and risk of getting prostate cancer. (The same paper I linked to in the previous sentence argues that the data strongly suggests obesity is a significant risk factor for prostate cancer death. This is based on observational epidemiological data with relative risks in the 1.25-range. Check out Part IV of our Studying Studies series for more on why we can’t necessarily rule out an effect, but we should be skeptical.)
One gripping headline and three paragraphs later, we learn that the study was done in mice. Not humans, or human cell lines. Plus, we learn that some prostate cancers lose a particular gene leading to the manufacturing of more fat (if you read the Nature Genetics article, this is expressed as “An SREBP-dependent lipogenic program is hyperactivated in prostate tumors from [Pten-null and Pml-null] mice”). There’s also a line about a fat-production-blocking obesity drug, but the context is unclear (i.e., how the investigators determined this—we’ll come back to this later).
Geneticists knew prostate cancers often start when a protective gene, PTEN, shuts down. But the tumors in men that lose only PTEN tend to languish, rarely spreading beyond the prostate and rarely becoming lethal.
The cancers change, though, if a second gene, called PML, also shuts down. Suddenly, indolent cells become cancers that spread and kill. But why?
In the new study, researchers found that when PML was lost, cancerous cells — in petri dishes and in mice — started churning out fat, which may protect the cells from certain toxic molecules. But the fat also may help the cancers spread, the researchers suggested.
How did the mice lose these particular genes (Pten and Pml)?2Genetic nomenclature note: Human gene symbols are written in all uppercase letters and italicized, whereas in mice only the first letter is uppercase and the rest are all lowercase (e.g., human PML and mouse Pml).
They never had them in the first place. The rodents from the study are called knockout (KO) mice, in which they are genetically modified so that an existing gene is “knocked out.” These particular mice are called genetically engineered mouse models (GEMMs) of prostate cancer (CaP).
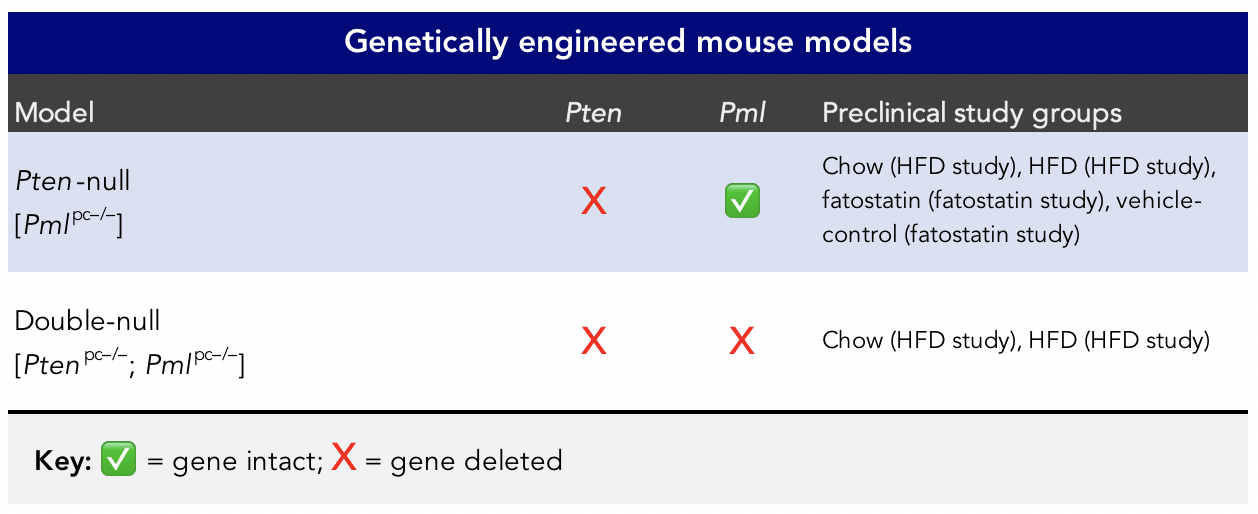
Figure 1. Genetically engineered mouse models of prostate cancer (Chen et al., 2018). These are called Probasin-Cre4 (Pb-Cre4) transgenic mice “maintained on a mixed C57BL/6 (80%) × 129S1/SvImJ (20%) background.”
The PTEN (i.e., phosphatase and tensin homolog) gene is considered to be a tumor suppressor gene and codes for the PTEN protein (naturally). The protein does several things, but notably for cancer, its useful for inhibiting the Akt signaling pathway. Akt is a protein kinase associated with tumor cell survival, proliferation, and invasiveness, so Akt inhibition is seen as a target for cancer therapy or prevention.
The PML (i.e., promyelocytic leukemia) gene is also considered as a tumor suppressor gene as it codes for the PML protein which is involved in blocking cell proliferation and inducing apoptosis (i.e., programmed cell death).
When Ms. Kolata alluded to CaP metastasis only if the mice “have a ready source of fat from the diet,” it was in the context of a “Pten-null” mouse (see Figure 1) with prostate cancer.
“Prostate-specific Pten inactivation leads to indolent, non-lethal invasive prostate cancer after long latency, while compound loss of Pten and Pml drives lethal and metastatic disease,” write the authors of the Nature Genetics paper (in a submission to an NIH data repository).
The implication is that the Pten-null mouse, with the Pml gene still intact and a ready source of fat from the diet, has prostate cancer that acts a lot like (i.e., similar phenotype) a double-null (i.e., double-KO) mouse that lacks both Pten and Pml. In other words, losing a gene that is frequently lost in prostate cancer (the authors estimated PTEN was lost in 14% of localized prostate cancer [LPC] and 66% of metastatic castration-resistant prostate cancer [mCRPC]), and coupling it with a high fat diet, produces a similar outcome as having a set of genetic mutations that are found in more aggressive prostate cancers.
Dr. Pandolfi has long tried to study prostate cancer spread in mice, but the rodents were not much help. Few genetic manipulations made prostate cancers spread in the animals as they do in humans.
Then one day, at a meeting with colleagues, Dr. Pandolfi had an idea: “What are the mice eating?” he asked. It was mouse chow, his co-workers said — a low-fat, vegetarian concoction.
“Why don’t we try a simple experiment?” Dr. Pandolfi recalled asking. “Why don’t we put our mice on a high-fat Western diet?”
It was the missing link. Mice with prostate cancers that had lost PTEN and that were fed a high-fat diet quickly developed tumors that grew rapidly and spread. It was as though fat in the diet had an effect similar to the loss of PML, the protective gene.
Some obvious questions:
- What is the “high-fat diet” (i.e., type and quantity of fat, type and quantity of carbohydrate and protein) in the study?
- How long was the study?
- How many mice in the study?
- How much fat (and other nutrients) did the mice consume overall?
- How much weight (and fat) did the mice gain?
- What were the metabolic changes in the mice?
- What percentage of the mice developed metastases?
- How did the “high-fat” diet (HFD) compare in these regards to the diet of their low-fat counterparts?
The HFD group in the study was fed the “Teklad Diet TD.06414” diet, consisting of 60% of kcals from fat. (A couple of notes: anytime we refer to “HFD,” we’re referring to this particular diet. Also, technically, one-thousand calories is equal to 1 kilocalorie, or 1 kcal for short. Here’s where it gets a bit tricky. Most people use the term “kilocalorie” and “calorie” interchangeably, including us sometimes. If that wasn’t confusing enough, Calories, with a capital “C,” is often utilized to make the distinction between calories with a lowercase “c.”)
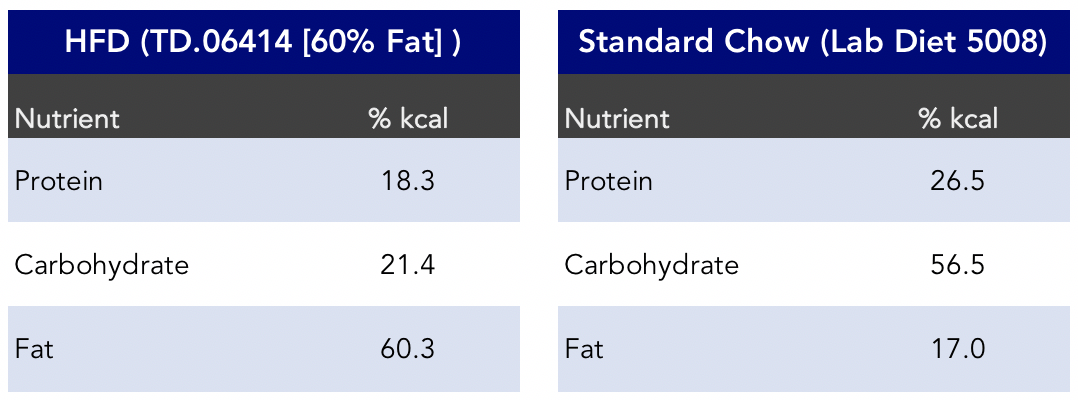
Figure 2. Macronutrient composition of the diets in the HFD study (Chen et al., 2018).
The HFD’s “key features” include “purified diet” and “diet-induced obesity.” The HFD by weight appears to contain just under seven times the amount of easily digestible sugars and more than three times the sucrose than the low-fat diet group by weight. Apparently, according to folks familiar with this diet, the mice won’t eat it without that much sugar in it. It seems the naturally herbivore mice didn’t evolve to find lard very palatable.
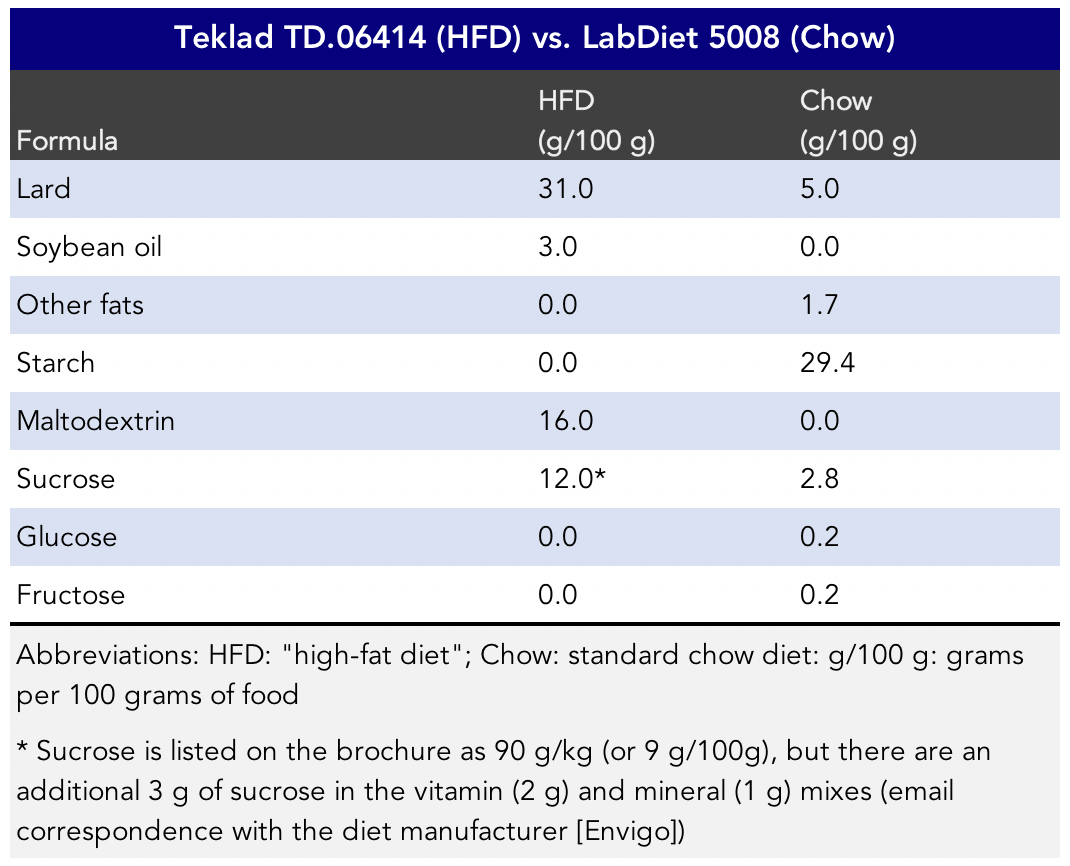
Figure 3. Comparison of the High-fat diet and chow diet ingredients (digestible carbohydrates and fats) in the HFD study (Chen et al., 2018).
The low-fat diet mice were fed LabDiet 5008. The “chow” diet is a relatively natural diet in terms of formula and is “a complete life cycle diet . . . paired with the selection of highest quality ingredients to assure minimal inherent biological variation in long-term studies,” according to the LabDiet brochure.
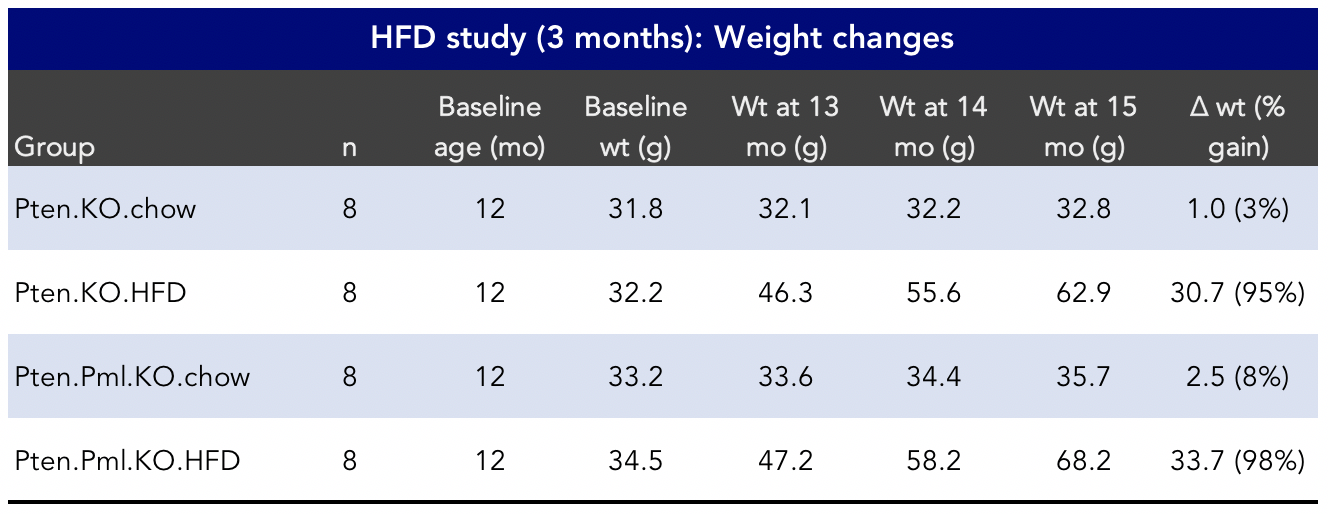
Figure 4: Weight changes in a preclinical high-fat diet (HFD) study (Chen et al., 2018).

Figure 5. Metastases in a preclinical high-fat diet (HFD) study (Chen et al., 2018).
Unfortunately, we don’t know how much fat, as well as carbohydrate and protein, nor do we know the metabolic impairment these mice endured on the diets because the authors did not monitor food intake or metabolic changes (the lead author of the article, Dr. Ming Chen is remarkably gracious to have several back-and-forth email correspondences with us in response to our questions).
In other words, while we know that the diet was 60% fat, we don’t know how much fat was consumed on the HFD or the standard chow because we don’t know how much they ate in absolute amounts. (If you asked me how much dietary fat I consumed yesterday, and I told you “60%,” I haven’t told you how much fat I consumed.)
Did you catch those body weights in the HFD groups in Figure 4? Both HFD groups gained an impressive amount of weight over the course of just three months.3Some of the weight gain could be due in part to the increases in tumor burden in these HFD groups, Dr. Chen noted in an email correspondence. Some of these mice more than doubled their body weights in 3 months.
To try to put this in perspective, if we extend this to humans, and use the NIH Body Weight Planner, to double an individual’s body weight, he should eat 12,302 kcals per day in order to reach his “goal” of 380 lbs in 90 days. If this person instead wanted to maintain his body weight of 190 lbs, he should eat 2,613 kcals per day, according to the Planner. In other words, his “intervention” is to eat just under five times (4.7) the number of kcals he should eat to maintain his body weight at baseline.

Figure 6. Estimated kcal consumption for an individual to double his body weight in 90 days using the NIH Body Weight Planner.
Mice are very different in regards to lifespan (about 40 times shorter) and metabolism (about seven times faster), so it’s hard to extrapolate these GEMM-studies to humans (which is kind of the point). A 48-hour fast in a rodent, for example, can lead up to 20% body weight loss. A human doubling his body weight in 3 months is fundamentally different than a mouse doing the same. (Three months may be the equivalent of 10 “mouse years” if we try to adjust for average lifespan.) But this is part of the point of this exercise. It’s difficult to intellectually compartmentalize these studies in animal models and appreciate these aren’t subtle differences when we read the newspaper headlines that lack context.
§
How much of a role are the other nutrients in the diet playing, particularly the type of carbohydrate used in “diet-induced obesity” diet formulas? It’s the Groundhog Day question of the Western diet and obesity (and its related diseases): what is it about the “Western diet” that seems to link up with obesity, type 2 diabetes, cancer, heart disease, stroke, Alzheimer’s disease, metabolic syndrome, and more?
One of the specific problems with the use of the term “high-fat diet” in research is the growing literature of “low-carbohydrate” or ketogenic diets and their association with cancer. For example, one study tested whether a no-carbohydrate ketogenic diet (NCKD) would delay prostate cancer growth relative to “Western” and “low-fat” diets in a xenograft model. They fed male SCID (severe combined immunodeficient) mice an 84% fat diet. After 24 days on their respective diets, all mice were injected with a cell line of human prostate cancer (LAPC-4 cells) and sacrificed when tumors approached a certain size.
The authors found that not only did they have to feed the NCKD more kcals than the Western and low-fat diets to equalize body weight between the groups (the low-fat diet was fed ad libitum [i.e., as much as desired] and the other arms were fed via a modified-paired feeding protocol), 51 days after injection NCKD tumor volumes were 33% smaller than Western diet mice and there was no difference between NCKD and low-fat mice. In addition, the longest survival of all groups was among the NCKD mice. In other words, these authors recognized that when mice were fed a “high-fat diet,” their cancer slowed and their lives lengthened, compared to diets lower in fat.
The same group of authors followed up with a study (Masko et al., 2010) in mice consuming not only an NCKD, but also a 10% carbohydrate (74% fat, 16% protein) or 20% carbohydrate (64% fat, 16% protein), and found the tumor volumes and overall survival on these diets to be similar to the 0% carbohydrate diet.
Notice the similarities in macronutrients between the 20% carbohydrate diet (Masko et al., 2010) and the Chen et al., 2018 HFD (Figure 7).
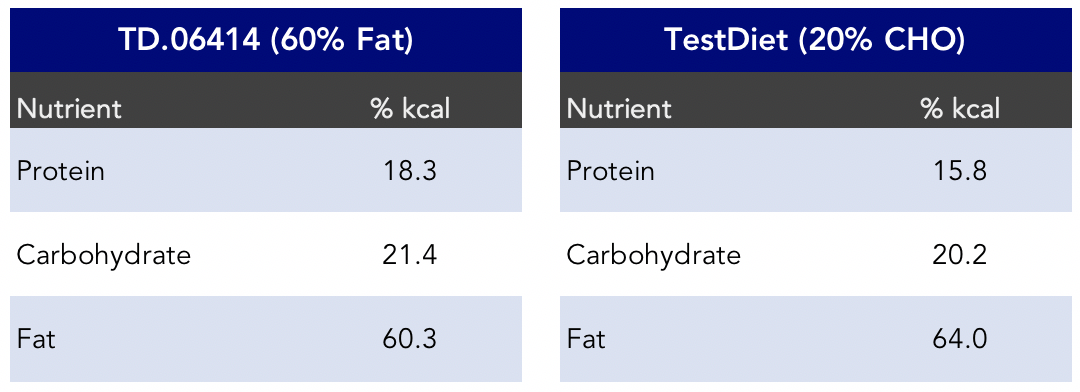
Figure 7. Macronutrient comparison between the TD.06414 HFD (Chen et al., 2018) and the TestDiet 20% carbohydrate diet (Masko et al., 2010).
How can these two diets seemingly produce divergent effects? One possibility is that it’s not an apples to apples comparison of study subjects. The mice were genetically different in the two studies. (Forgive me for being skeptical, but if we’re going to question the applicability of one mouse model to another mouse model, perhaps we should also question the applicability of mouse models to other species, such as humans?) An additional possibility is that the TestDiet formula did not contain any easily digestible sugars while the TD.06414 was relatively high in maltodextrin and sucrose (Figure 8) and that may have affected the outcomes.
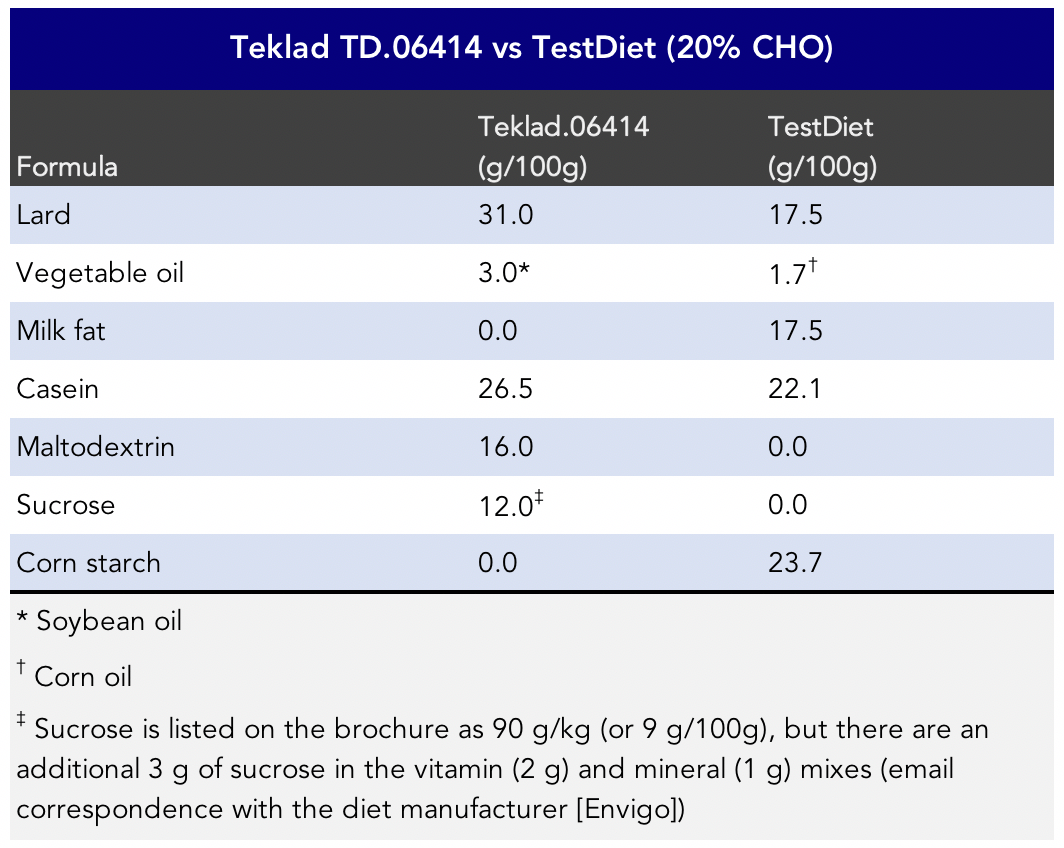
Figure 8. Formula comparison between the TD.06414 HFD (Chen et al., 2018) and the TestDiet 20% carbohydrate diet (Masko et al., 2010). Abbreviations: CHO: carbohydrate.
A recent systematic review of tumor cell growth and survival time with the ketogenic diet in animal models concluded that recent findings have shown that ketogenic diets might be able to inhibit malignant cell growth and increase survival. Taken together, the evidence suggests “high-fat” diets need more context, specifically the quantity and type of carbohydrates.
In other words, is dietary fat per se, the promoter of more aggressive prostate cancer? It depends, may be the responsible answer. Jeff Volek’s group, for example, have shown that the amount of fat that accumulates in the blood is linked to the amount of carbohydrates consumed. “Having a lot of saturated fat in your body is not a good thing,” said Volek. “The question is, what causes people to store more saturated fat in their blood, or membranes, or tissues?”
Is dietary fat helping to fuel the spread of prostate cancer or is there more to the HFD diet than just fat, leading to metabolic impairment, lipid accumulation, and metastases? Unfortunately, you’ll recall, we don’t know what the metabolic changes were in the Nature Genetics study. (We do know the “key findings” in the promotional brochure of the “HFD” used in the study [TD.06414] show “impaired glucose tolerance . . . hyperinsulinemia, and . . . increased liver accumulation of lipids.”)
§
Then the group asked a bigger question: Could they could protect mice from metastatic cancer by blocking fat production? That led to the experiment with a new obesity drug, fatostatin. It not only halted the cancer’s spread in the animals, but made it regress.
In a separate experiment, this “obesity drug,” called fatostatin (who comes up with these names?), was given to Pten and Pml KO mice along with a “standard chow” diet (according to the lead author of the study, Dr. Chen, via email correspondence) for 2 months.

Figure 9. Preclinical 2-month study of fatostatin in double-null mice (Chen et al., 2018). None of the six mice on fatostatin had cancer that metastasized while two of the six mice in the “vehicle control” of corn oil had metastases develop.
The mice treated with fatostatin were fed the standard chow diet (17% fat, 27% protein, 57% carbohydrate). If fatostatin does indeed prevent prostate cancers in mice from spreading, we don’t necessarily know it’s because the drug is blocking fat production. The statins that most of us are familiar with (i.e., HMG CoA reductase inhibitors) lower serum cholesterol and can help prevent cardiovascular (CVD) events, but to say that statins prevent CVD events because they lower serum cholesterol would be at best an oversimplification of how statins work.
There’s also the lingering question: why didn’t the investigators include mice on the HFD in this trial? (It was also noted in the Times article, these same investigators are planning a clinical trial with fatostatin to treat prostate cancer in humans.)
§
If there’s one thing to take away from this exercise, it’s to understand that context matters.
Take the external and internal validity into context and remember what questions we’re asking here.
For the HFD study, what is the degree to which the results would hold true for men with localized prostate cancer, including the loss of PTEN and/or PML, eating nothing but unlimited cheesecake-protein-shakes blended in Coca-Cola with added butter on top (Figure 10), indefinitely? 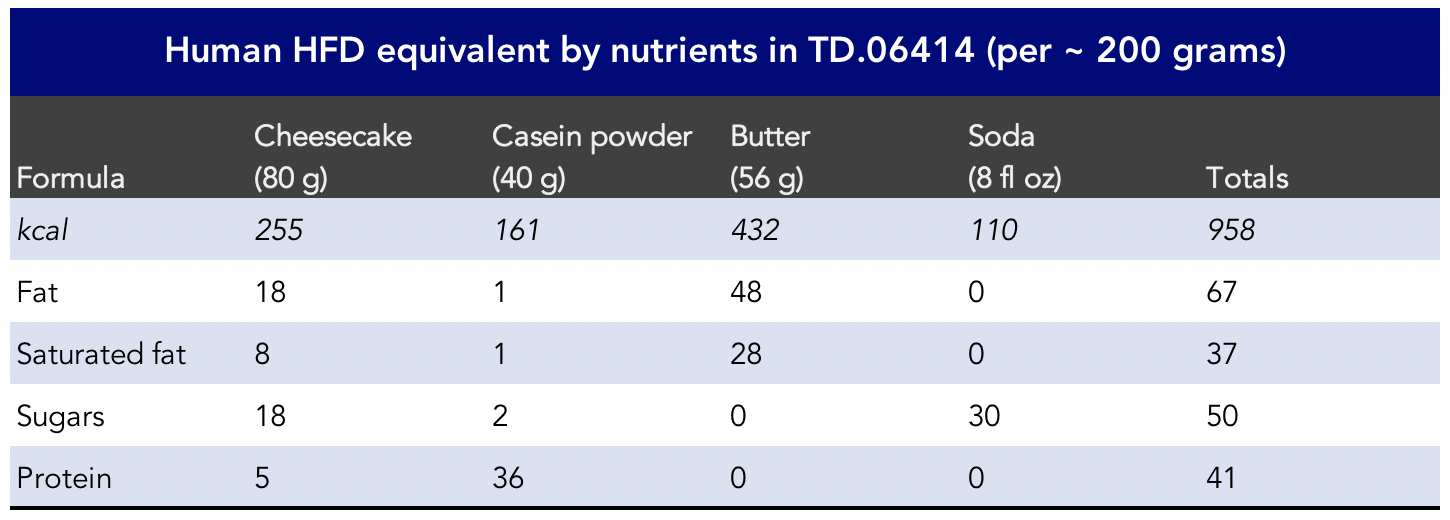
Figure 10. Peter’s Cheesecake bomb recipe. The macronutrients (and sugars) are used to mimic the TD.06414 used in the Chen et al., 2018 study.
One thing I haven’t discussed yet is that the percentage of cases (according to sample analyses by Chen et al., 2018) the PTEN gene is lost in localized prostate cancer (LPC) is in the neighborhood of 14%. For metastatic castration-resistant prostate cancers (mCRPCs), it occurs in about 66% of samples. (Estimated incidence of mCRPC in 2009 was 36,100. Metastasis occurs in approximately 5% of prostate cancer cases [Figure 12].)
For the fatostatin study, what is the degree to which the results would hold true for men with localized prostate cancer, including the loss of PTEN and PML eating nothing but a 17% fat (56% carbohydrate) diet with less than 30 grams of sugars (likely less than five times the average American consumes in caloric sweeteners), while on a fatostatin, indefinitely?
For both the HFD and fatostatin study, what are we to make of the diets and metabolic impairment if food intake and metabolic changes were not monitored?
Studying the effects of diets on genetically engineered mouse models of cancer with genes knocked out is not the same thing as studying the effects of diets on humans. The Times headline (High-Fat Diet May Fuel Spread of Prostate Cancer”), the Nature Genetics title (“An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer”), and the press release from Beth Israel (“Flipping the Switch: Dietary Fat, Changes in Fat Metabolism May Promote Prostate Cancer Metastasis”), all managed to omit the ending: “. . . in mice.”

Figure 11. Context matters.
The results of these studies are not generalizable to the 99% of the population that does not have prostate cancer and it’s questionable if they are even generalizable to the 1% that do. If you recall, PTEN is lost in 14% and 66% of localized and mCRPC cases, respectively, which may knock the percentage down to 0.1% of the population that’s affected.
For the fatostatin study, conducted only in co-deleted (Pten and Pml) animal models, generalizability takes a bigger hit: estimated LPC with co-deletion in the human population was zero (none of the samples were co-deleted), and 20% for mCRPC. In other words, the animals used in the fatostatin study “represent” about 7,000 men a year, or the 0.002% (1-in-50,000) of the US population who are estimated to have mCRPC and co-deletions of PTEN and PML.
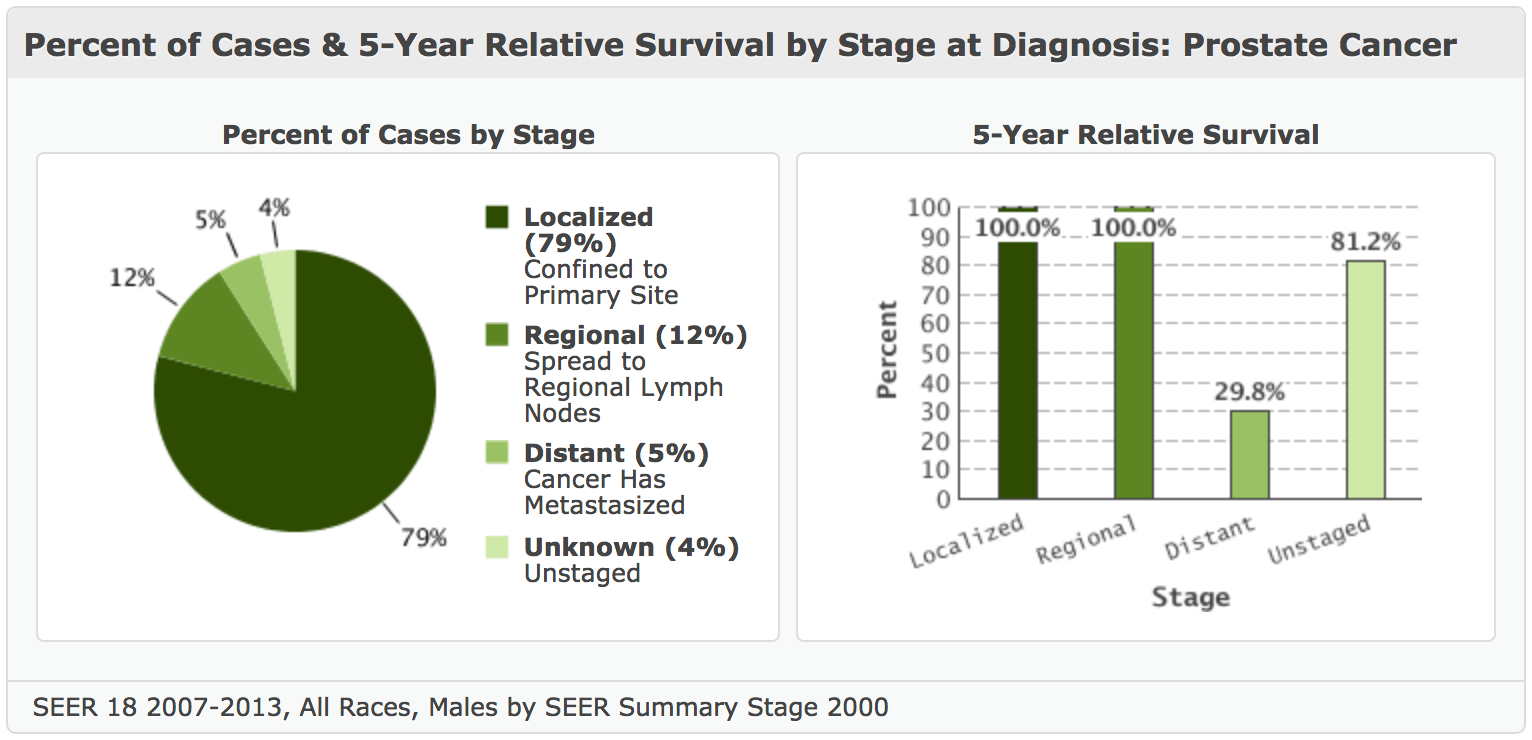
Figure 12. Prostate cancer: percent of cases by stage and 5-year relative survival rates in the US. National Cancer Institute.
It’s actually pretty easy to miss the broader memo even though it was contained in the three aforementioned headlines above: the study pertained to animal models (well, this part was left out) who already had prostate cancer and whether the diet could promote metastasis thereafter. (It looks like the Economic Times did not get the memo: “Eating cheeseburgers and fries can you make you vulnerable to prostate cancer,” the headline reads.)
A “high-fat diet” is not simply a high-fat diet. Specifying the level and type of carbohydrates and protein matter. People have done the research showing this and it shouldn’t be ignored (or missed).4This is a broader issue that probably needs to be addressed: with so much information out there and so many different groups working on these topics, it’s difficult for researchers to even know what all of the “known-knowns” are in their own field. This matters not only for the tens of a percent of men with prostate cancer coupled with the loss of PTEN, but also for the general population.
There’s an epidemic of poor terminology, lack of consistency, lack of communication, and lack of a rudder pervading the field of clinical and observational studies on diet and it translates to confusion for everyone (the field, the investigators, the media, the public) involved. I wouldn’t quibble over semantics if it wasn’t relevant to the big picture. More than 1600 people die each day from cancer (about 73 from prostate cancer). There are serious unintended consequences of being less than hypervigilant about how we communicate and interpret studies.



