
Millions of Americans schedule an annual physical health examination. During their visit, many can expect to get their blood drawn for a standard lipid panel, which ultimately provides the patient with a readout of total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and triglycerides (TGs). What virtually nobody among them will get is an apolipoprotein B (apoB) test. Yet this is considered the ideal, if not essential, measurement to assess lipoprotein-related atherosclerotic cardiovascular disease (ASCVD) risk, according to recent guidelines and an illuminating review by Dr. Allan Sniderman and his colleagues. In order to appreciate why this matters—and before digging into Sniderman et al.’s review—you need to understand some of the basics of lipids and lipoproteins first.
If it seems like what follows is “too much,” consider that we are talking about the most prevalent disease in the developed world, and consider, at least for a moment, how much misinformation exists around the ideas of cholesterol.
“We must admit that our opponents in this argument have a marked advantage over us. They need only a few words to set forth a half-truth; whereas, in order to show that it is a half-truth, we have to resort to long and arid dissertations.”
― Frédéric Bastiat, Economic Sophisms, 1848
Lipids are a class of molecules that are not soluble in water and therefore cannot circulate in plasma (the liquid flowing through our arteries and veins), as plasma is 90% water, any more than you can dissolve a tablespoon of olive oil in a glass of water. A lipoprotein is essentially a vehicle that carries these lipids through plasma. A lipoprotein is like a submarine that transports cargo, with the bulk of the cargo consisting of TGs and cholesteryl ester (CE), which is cholesterol with one fatty acid attached to it. The 360-degree hull of this submarine consists of a one-molecule thick layer composed mainly of phospholipids, “free” cholesterol (meaning it’s not bound to a fatty acid), and proteins called apoproteins or apolipoproteins, which wrap around the exterior of the lipoprotein. Apolipoproteins are the “protein” in lipoprotein. This metaphorical submarine is not cigar-shaped like those found in our oceans, but is ball-shaped or spherical found in our plasma, as you can see in Figure 1.
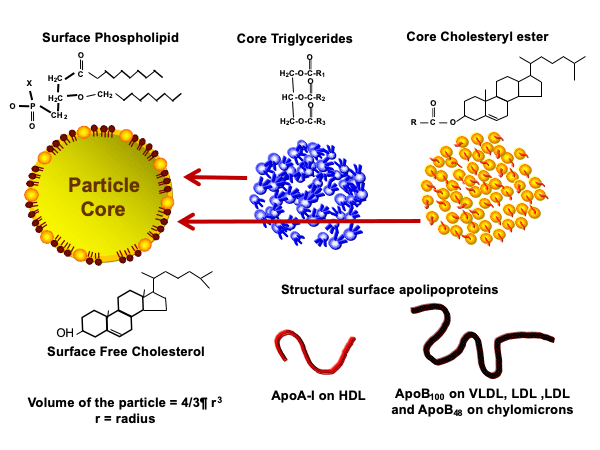
Figure 1. Component parts of a lipoprotein.
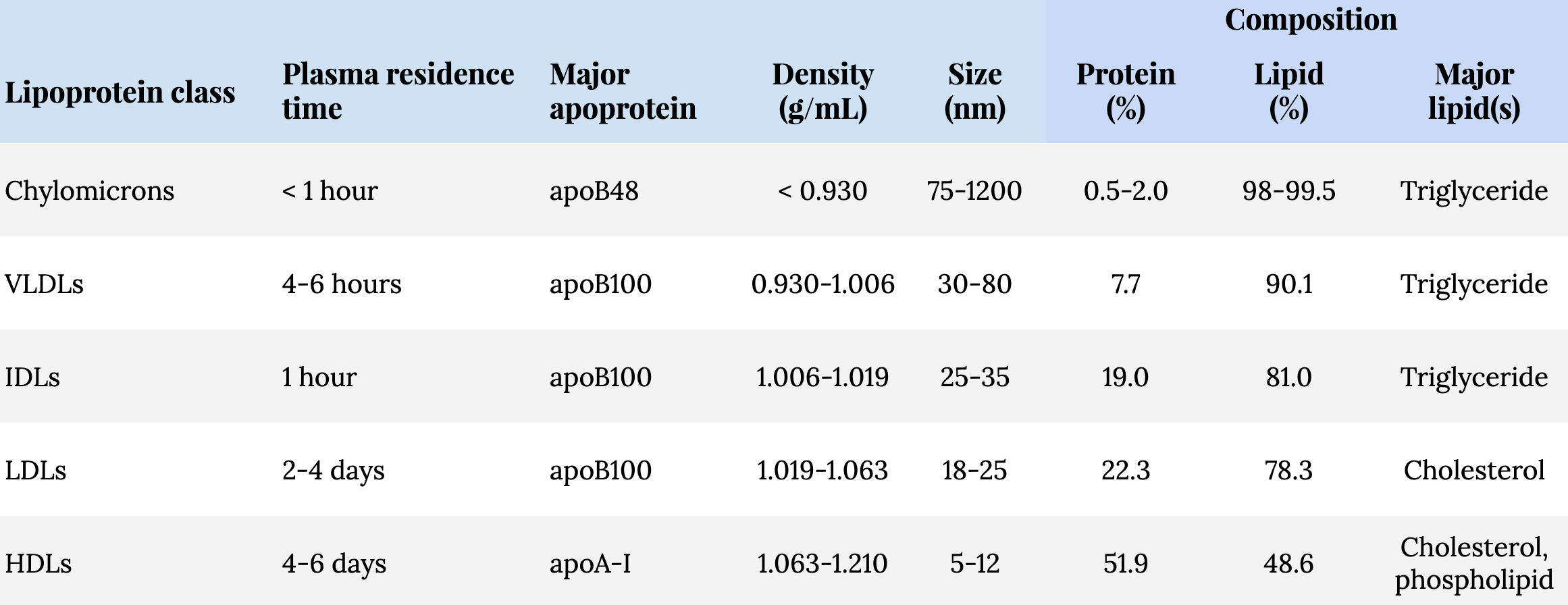
The density of lipoproteins were discovered many decades ago by drawing blood into a sealed collection tube, spinning the living daylights out of that tube, a process called ultracentrifugation, and seeing where they settled. Most lipoproteins then became classified by their density. The lipoproteins that sit in the plasma at the top of the tube have the lowest density, and these lipoproteins are called chylomicrons (CMs), which are produced in the small intestine. The other four major classes of lipoproteins are all produced in the liver. In order of density, lowest to highest, they are: very-low-density lipoproteins (VLDLs), intermediate-density lipoproteins (IDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs). Among the LDL class, there is a related lipoprotein called lipoprotein “little a,” or Lp(a) for short. The density of the particles are related to their lipid-to-protein ratio. The particles with the most lipids, like CMs and VLDLs, are larger and more buoyant (and therefore less dense) compared to particles carrying fewer lipids, like LDLs and HDLs, which are smaller and denser. I think it’s easier to understand this if you consider the information and illustration in Table 1 and Figure 2, respectively, putting the size, density, and composition of the different classes of lipoproteins into context.
Table 1. Classes and characteristics of lipoproteins.1The major apoprotein in chylomicrons are referred to as apoB48, while those produced in the liver are apoB100. The numbers represent the different molecular weights between the two. The apoB produced in the intestine is a truncated version of the hepatically produced apoB100: The intestinal variant has 48% (i.e., apoB48) of the molecular weight of the apoB produced in the liver (i.e., apoB100).
Lipoproteins are not only classified by their density, but laboratory techniques allow for the measurement of their apolipoproteins. While there are at least 10 classes, and several subclasses, of apolipoproteins, our focus here is on apolipoprotein B (apoB).
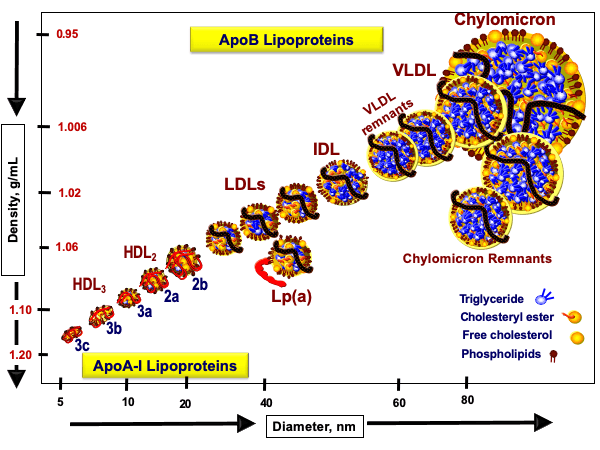 Figure 2. Size, density, and apolipoprotein identification of lipoproteins.
Figure 2. Size, density, and apolipoprotein identification of lipoproteins.
It turns out that each CM, VLDL, IDL, LDL, and Lp(a) has one, and only one, apoB per particle. This allows apoB measurement to serve as a particle concentration for these lipoproteins.2The ApoA-I-containing family of particles belongs to the HDLs, but each HDL particle carries a variable number (i.e., 1-5) of copies. All of the lipoproteins in Figure 2, with the exception of HDLs, carry a single copy of apoB. In other words:
Total apoB concentration = CM-apoB + VLDL-apoB + IDL-apoB + LDL-apoB + Lp(a)-apoB
Because of its extended plasma residence time, more than 90% of apoB-containing particles in plasma are LDLs, which means apoB measurement mostly represents LDL particle concentration. Overall, you can think of apoB as a collective measure of all potentially atherogenic lipoproteins including, if present, Lp(a), and this entire family of apoB-containing lipoproteins are shown in Figure 3, including VLDL and its atherogenic remnants.
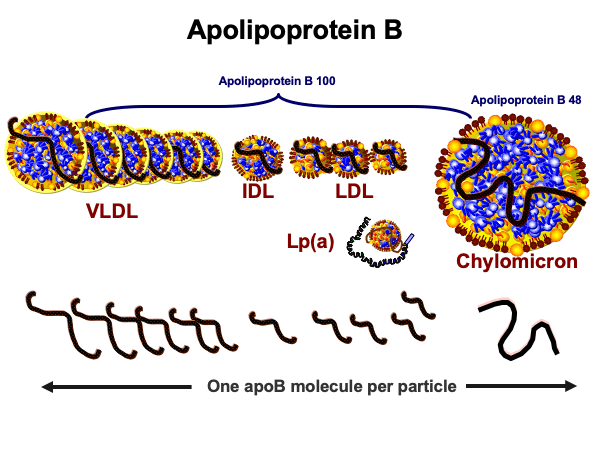
Figure 3. Apolipoprotein B-containing lipoproteins.
When you get a standard lipid panel and the familiar readout of TC, LDL-C, HDL-C, and TG, it’s showing you the mass of these lipids (i.e., cholesterol, except in the case of the TG measurement) per unit volume of plasma. Here in the States, the mass is shown in milligrams and the unit volume of plasma is a deciliter (i.e., one-tenth of a liter). For example, an assay may show the mass of TGs are 100 mg/dL and the mass of cholesterol in all of the lipoproteins circulating per unit volume of plasma is 200 mg/dL. In other words, TC is the sum of all of the cholesterol within each of the distinct lipoprotein classes, as illustrated in Figure 4 and is expressed in the following equation:
TC = VLDL-C + IDL-C + LDL-C + Lp(a)-C + HDL-C
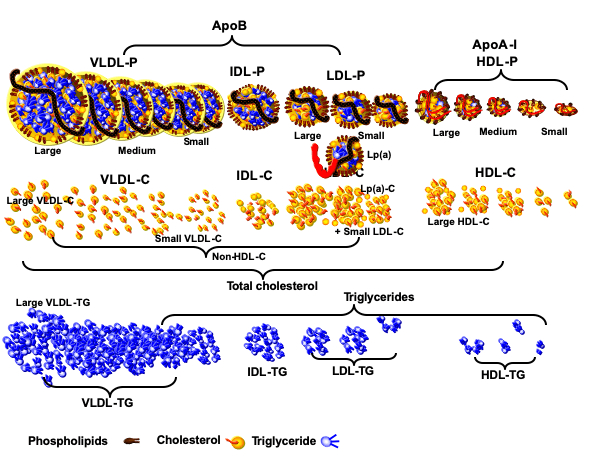
Figure 4. Lipoproteins and their cargo content.
Although LDL-C can be directly measured, LDL-C reported by most labs is usually calculated based on direct measurements of TC, HDL-C, and TG, and is called the Friedewald formula. This formula subtracts HDL-C and VLDL-C from TC to estimate LDL-C. VLDL-C is estimated as TG/5, which assumes VLDLs have 5 times more TG than cholesterol.3Unfortunately, that assumption is often erroneous especially when TG concentrations escalate above 130-150 mg/dL. Historically a normal TG has been listed as 150 mg/dL which would make a normal VLDL-C 30 mg/dL. Since a TG of 150 mg/dl is the 50th-75th percentile population cutpoint, desirable VLDL-C should be much lower (likely 10-15 mg/dL).
Therefore LDL-C is calculated as:
Calculated LDL-C = TC – HDL-C – (TG/5)
The non-HDL-C measurement, shown in Table 2, is a metric that is increasing in popularity for predicting ASCVD risk. By subtracting HDL-C from TC, the resulting non-HDL-C is the sum of all cholesterol carried within the apoB-containing lipoproteins. Non-HDL-C measurement is to apoB measurement what direct LDL-C measurement is to LDL particle measurement.
Table 2. Commonly reported lipid concentration measurements.
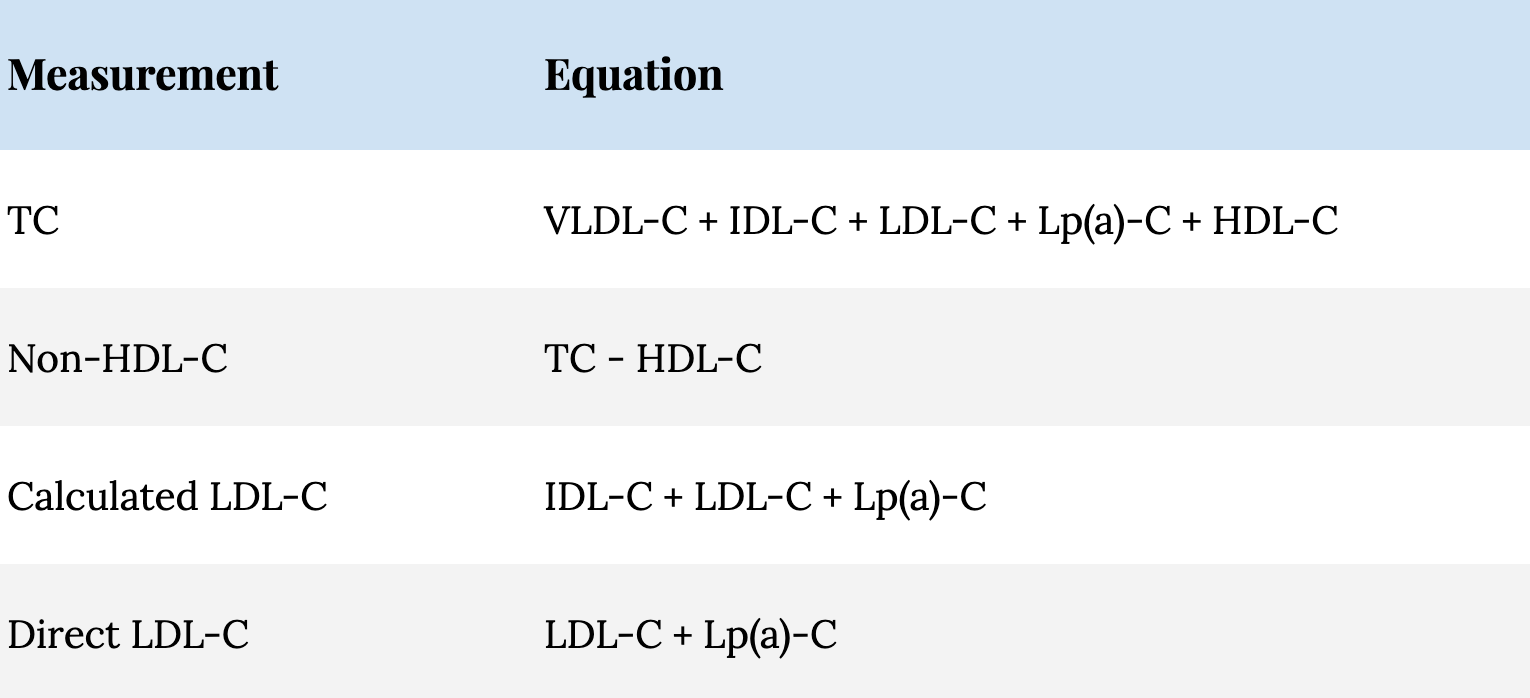 So what is the upshot of this Lipidology 101?
So what is the upshot of this Lipidology 101?
- Cholesterol (and triglycerides) can’t just float around in our circulation since they are not water-soluble, so we need elaborate vehicles to move them around. These vehicles are called lipoproteins.
- Lipoproteins differ by many properties, but a couple stand out: their density and their apoproteins. On the one hand, we have a family of high-density lipoproteins, cloaked in apoA-I apoproteins, and on the other hand, we have a family of low-density lipoproteins, cloaked in apoB apoproteins.
- The apoB-bearing lipoproteins are the atherogenic ones, and in most people ~90% of the apoB-bearing lipoproteins are low density lipoproteins (LDLs).
- The common, or typical, lipid measurements used by most physicians (e.g., LDL-C, non-HDL-C) are simply correlates of apoB particle concentration. Importantly, these surrogates can often be misleading.
In Part 2 of this email, we’ll discuss why this discordance between apoB and its surrogates exists and why it poses a problem for anyone interested in reducing their risk of atherosclerotic heart disease.
§
In a recent review article in JAMA Cardiology, Allan Sniderman and his colleagues make the case that apolipoprotein B (apoB) level—rather than LDL-C, non-HDL-C, or even LDL particle count (LDL-P)—is the best measure of potentially atherogenic lipoproteins.
Important in understanding why lipidologists like Dr. Sniderman are increasingly arguing for more direct measurements of the number of lipoproteins themselves, rather than the cholesterol or triglycerides contained within them, is the concept of concordance and discordance. The most common scenario in which we can examine this concordance/discordance split is contrasting LDL-C (the commonly used measure of cholesterol contained within LDL) and LDL-P (the less commonly used measure of the number of LDL particles).
A high level of LDL-C predicts a high level of LDL-P. Same for low LDL-C predicting low LDL-P. When LDL-C and LDL-P agree, they are concordant, and they equally predict the same outcome: both are high (or low) relative to the population. When they disagree—one is high and one is low—they are discordant. (Note: we’re not talking about comparing the absolute numbers of either metric—they are measuring different things in different units—we’re talking about how each stacks up to their respective population mean.)
When LDL-C is concordant with LDL-P, LDL-C is a fine predictor of adverse cardiovascular events. But when there’s discordance, it’s imperative to know which—LDL-C or LDL-P—is the correct predictor. To examine this question we turn to two of the largest studies comparing subjects over time using “hard” outcomes, the Framingham Offspring and MESA cohorts. Start with Figure 5 (the relatively homogeneous Framingham Offspring population) and pay special attention to the individuals that had the lowest event-free survival (the blue and yellow curves that fall the fastest). They had low or high LDL-C, but universally high LDL-P. Conversely, look at the individuals with the highest event-free survival (the red and green curves that fall the slowest). They had high or low LDL-C, but universally low LDL-P.
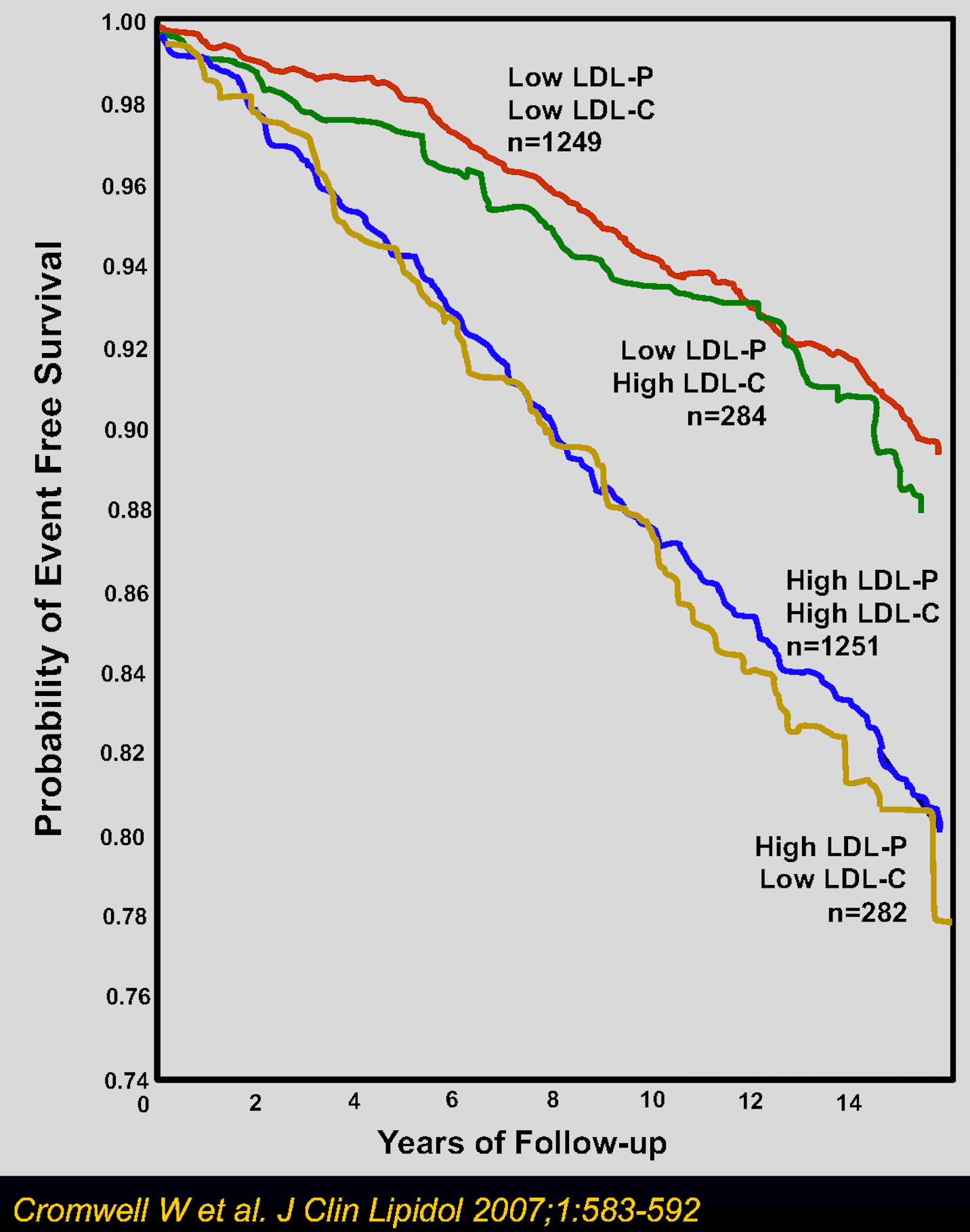 Figure 5. Event-free survival among participants with LDL cholesterol (LDL-C) and LDL particle number (LDL-P) above or below the median. Median values were 131 mg/dL for LDL-C and 1414 nmol/L for LDL-P.
Figure 5. Event-free survival among participants with LDL cholesterol (LDL-C) and LDL particle number (LDL-P) above or below the median. Median values were 131 mg/dL for LDL-C and 1414 nmol/L for LDL-P.
If you want to do the same exercise, but in reverse (i.e., look at the rate of events occurring rather than freedom from events), and do so in a much more heterogeneous population, look at the cumulative incidence of events in the Multi-Ethnic Study of Atherosclerosis (MESA) population (Figure 6). The exact same pattern emerges: high LDL-P (independent of LDL-C) predicts more events (the red and yellow curves), while low LDL-P (independent of LDL-C) predicts fewer events (the black and blue curves). This point is not subtle and it’s one of the few times when “eyeball statistics” do the trick—your eyeballs tell you the answer even if you don’t know the difference between a Cox proportional hazard ratio, a Fisher’s exact test, and a box of Lucky Charms.
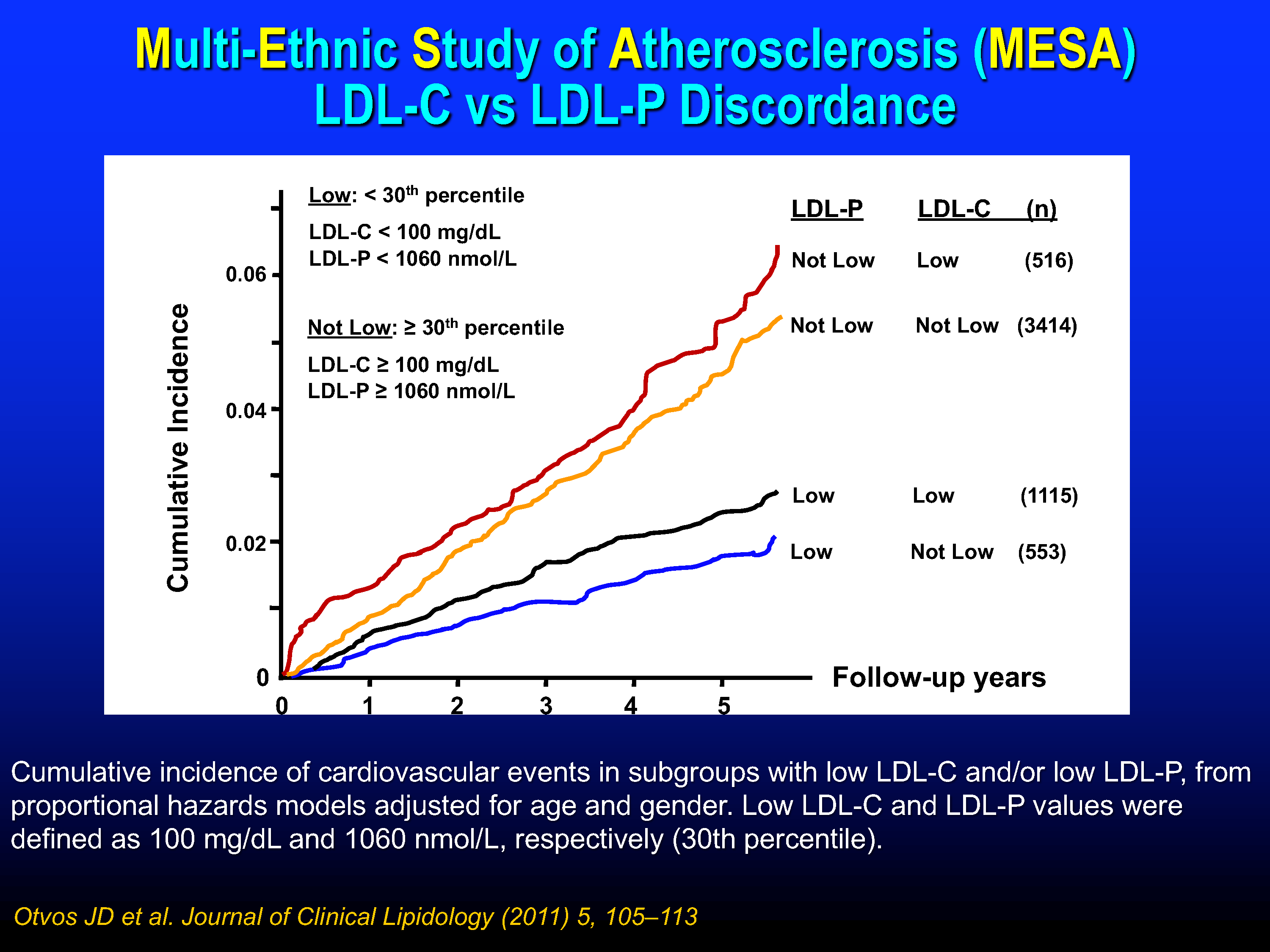 Figure 6. Cumulative incidence of cardiovascular events stratified by concordance and discordance.
Figure 6. Cumulative incidence of cardiovascular events stratified by concordance and discordance.
This point is critical in determining patient risk for atherosclerotic cardiovascular disease (ASCVD), argues Sniderman et al. (One subtle distinction is that the Sniderman paper refers to the frequent discordance between non-HDL-C levels and apoB levels.)4More technically, it’s apoB-containing particle levels. When I refer to apoB levels or apoBs, I’m talking about the concentration or number of lipoproteins that have an apoB attached to it.
An important question you are probably asking is what drives this discordance between the cargo of the particle and the number of particles? The composition and sizes of apoB-containing particles are not set in stone. They vary from person to person and particle to particle, depending on the person’s metabolic health and genetic makeup. For example, an LDL carrying relatively more TG (so-called“TG-rich”) contains less cholesterol (so-called “cholesterol-depleted”). Also, a smaller diameter LDL leaves far less space within its core to carry cholesterol, regardless of TG content. If you have a lot of smaller, cholesterol-depleted LDLs, this is a recipe for discordance: low LDL-C and high LDL-P. On the other hand, if you have a lot of larger, cholesterol-rich particles, this is also a recipe for discordance, but in this case you’re more likely to have higher LDL-C relative to lower LDL-P.
What actually drives these changes in size and composition of apoB-containing particles from a metabolic perspective is a very important topic that we’ll explore in the future.
While I have historically used LDL-P and apoB metrics somewhat interchangeably, one advantage of measuring apoB over LDL-P is that apoB encompasses all of the potentially atherogenic particles: not just LDLs, but VLDLs, Lp(a), IDLs, and potentially chylomicrons. And this point has me relying more and more heavily on apoB today than on LDL-P in my patients and myself.
Why are particle concentrations better at predicting risk than cholesterol concentrations? Sniderman et al.’s hypothesis of atherogenesis is relatively straightforward and essentially comes down to numbers and time course: A greater number of apoB-containing particles leads to a greater number of these particles that enter and get trapped within the wall of the artery, leading to a greater amount of injury to the arterial wall. It’s somewhat stochastic: the more shots on goal (particles hitting the artery wall), the more chances of goals (particles getting through the cracks).
While the inevitableness of trapped particles within our arterial walls may sound depressing, this mechanism of atherogenesis offers at least a partial solution: lower the number of apoB-containing lipoproteins. If apoB-containing particles are the basic unit of injury and more particles means more problems, let’s see what it looks like when we reverse this line of thinking: A smaller number of apoB-containing particles leads to a smaller number of these particles that enter and get trapped within the wall of the artery, and therefore less injury to the arterial wall.
Beyond lifestyle—a term I despise, but I think you understand what I’m referring to: smoking cessation, blood pressure control, correcting insulin resistance, and more—we know that lipid-lowering drugs like statins, ezetimibe, PCSK9 inhibitors, and bile sequestrants all lower apoB through mechanisms that we can explore later. For people at moderate to very-high risk with high apoBs, interventions like these can help lower that risk. However, the only way for a clinician to know when apoB and cholesterol measurements are concordant or discordant is to measure apoB and compare it to the lipid profile. In virtually all trials when discordance is present, ASCVD-risk better follows the apoB. If they are correct, and apoB is our best measure of risk, this makes for a very compelling case for including apoB measurement—and perhaps even replacing LDL-C and non-HDL-C altogether—in clinical care.
Bottom line: while there is more to ASCVD risk than just lipoproteins, if you don’t know your apoB level, you are not fully taking advantage of the tools at our disposal to estimate your risk of cardiovascular and cerebrovascular disease. Imagine a smoker not knowing they smoke, or someone with high blood pressure never getting strapped up to a BP cuff. Know your risks and manage them accordingly.
– Peter



