Allan Sniderman is a highly acclaimed Professor of Cardiology and Medicine at McGill University and a foremost expert in cardiovascular disease (CVD). In this episode, Allan explains the many risk factors used to predict atherosclerosis, including triglycerides, cholesterol, and lipoproteins, and he makes the case for apoB as a superior metric that is currently being underutilized. Allan expresses his frustration with the current scientific climate and its emphasis on consensus and unanimity over encouraging multiple viewpoints, thus holding back the advancement of metrics like apoB for assessing CVD risk, treatment, and prevention strategies. Finally, Allan illuminates his research that led to his 30-year causal model of risk and explains the potentially life-saving advantages of early intervention for the prevention of future disease.
Subscribe on: APPLE PODCASTS | RSS | GOOGLE | OVERCAST | STITCHER

We discuss:
- Problems with the current 10-year risk assessment of cardiovascular disease (CVD) and the implications for prevention [4:30];
- A primer on cholesterol, apoB, and plasma lipoproteins [16:30];
- Pathophysiology of CVD and the impact of particle cholesterol concentration vs. number of particles [23:45];
- Limitations of standard blood panels [29:00];
- Remnant type III hyperlipoproteinemia—high cholesterol, low Apo B, high triglyceride [32:15];
- Using apoB to estimate risk of CVD [37:30];
- How Mendelian randomization is bolstering the case for ApoB as the superior metric for risk prediction [40:45];
- Hypertension and CVD risk [49:15];
- Factors influencing the decision to begin preventative intervention for CVD [58:30];
- Using the coronary artery calcium (CAC) score as a predictive tool [1:03:15];
- The challenge of motivating individuals to take early interventions [1:12:30];
- How medical advancement is hindered by the lack of critical thinking once a “consensus” is reached [1:15:15];
- PSK9 inhibitors and familial hypercholesterolemia: two examples of complex topics with differing interpretations of the science [1:20:45];
- Defining risk and uncertainty in the guidelines [1:26:00];
- Making clinical decisions in the face of uncertainty [1:31:00];
- How the emphasis on consensus and unanimity has become a crucial weakness for science and medicine [1:35:45];
- Factors holding back the advancement of apoB for assessing CVD risk, treatment, and prevention strategies [1:41:45];
- Advantages of a 30-year risk assessment and early intervention [1:50:30];
- More.
Get Peter’s expertise in your inbox 100% free.
Sign up to receive An Introductory Guide to Longevity by Peter Attia, weekly longevity-focused articles, and new podcast announcements.*Pre-show notes*
- Allan is a senior scientist at the Research Institute Of McGill University Health Center and the Edwards Professor of Cardiology and Professor of Medicine at McGill University
- He’s the director of the Mike Rosenbloom Laboratory for Cardiovascular Research at Royal Victoria Hospital in Montreal
- He was elected a fellow of the Royal Society of Canada in 2009
- He’s also been an enormous mentor for Peter over the past 10 years; he’s certainly, one of the 3 or 4 people I would count that has nearly single-handedly provided him with the education that he uses to help understand cardiovascular disease
- Peter has included him in some of the things I’ve written about. And also he’s been mentioned a number of times on previous podcasts featuring no less than Tom Dayspring and Ron Krauss
- His memberships are probably too numerous to mention, but a few of them include the Royal College of Physicians and Surgeons in Canada, the American College of Cardiology, the American College of Physicians, the Canadian Cardiovascular Society, the American Society for Clinical Investigation, the American Federation for Clinical Research and a number of others
- In this episode we go very deep on cardiovascular medicine
- Our discussion doesn’t begin in the way I intended, in the first 10 or 15 minutes, Allan lays out some of the most clear, yet complex concepts that you’ll ever hear anybody talk about with respect to apoB and risk management
- Don’t be dissuaded by how complicated the first 10-15 minutes of this podcast are
- Instead, sit tight and this episode will walk you through the journey of what apoB is and why it is a superior metric for predicting risk of atherosclerosis relative to the far more commonly used metric, LDL cholesterol
- And the metric that is better than LDL cholesterol, but still inferior to apoB, which is non-HDL cholesterol
- This episode will also go into great detail about the role of triglycerides, HDL cholesterol, total cholesterol, et cetera, in the understanding of and prediction of cardiovascular disease
- This episode will get into some of the interesting exceptions, i. e., disease states where not knowing apoB poses an enormous blind spot
- This episode will talk about the challenges that face people like Allan, as they try to disseminate the most leading edge and cutting edge science on this topic in an environment that is really wed to guidelines and consensus-based recommendations that don’t necessarily incorporate all of the best evidence
- This episode will explain some concepts like Mendelian randomization and how that’s been used to further bolster the case for apoB as the superior metric for risk prediction
- Coronary artery calcium scoring will also be discussed
- This is obviously an important tool used by many physicians, and it’s important to understand how it’s useful and what its blind spots are
Problems with the current 10-year risk assessment of cardiovascular disease (CVD) and the implications for prevention [4:30]
Understanding atherosclerosis/ cardiovascular disease
- Peter has referenced books Allan gave him numerous times
- ApoB in Clinical Care by Jacqueline de Graaf, Patrick Couture, and Allan Sniderman
- Atlas of Atherosclerosis Progression and Regression (Encyclopedia of Visual Medicine Series) by Herbert C. Stary (2003)

Figure 1. Book cover Herbert Stary’s Atlas of Atherosclerosis Progression and Regression showing plaque rupture “rings”. Image credit: Amazon.com
- Peter asks him why he gave him this last book
- Atherosclerosis, it’s a disease in the tissue, yet almost everything that lipid people talk about is in plasma
- One must understand the natural history of the disease to learn how to construct a strategy to prevent it
- Although much of Allan’s work has been on apoB, the more important part has been on understanding how the natural history of atherosclerosis should direct a prevention strategy
- What this leads to is that every major guideline in the world bases their selection of subjects for statin prevention on the ten-year risk of disease
- This was a huge step forward in 1980 and 1990
- But it fundamentally makes prevention of premature disease almost impossible
- What this leads to is that every major guideline in the world bases their selection of subjects for statin prevention on the ten-year risk of disease
Problems with the 10-year risk approach
- When one plugs in the numbers to calculate a patient’s risk for any of the risk algorithms, one expects the output to be the patient’s risk of cardiovascular disease (CVD) but it isn’t
- The American College of Cardiology ASCVD Risk Estimator Plus
- The calculation is driven by the age and sex of the patient
- Things like cholesterol and blood pressure contribute minimally to the actual calculation of ten-year risk
- So if the patient is 35, there isn’t a risk calculator for them
- If the patient is 40, everyone’s risk is low at age 40
- It isn’t until age 55 or 60 that the risk gets over the threshold for the American Prevention Guideline treatment
- So prevention really starts at 55 to 60, but almost half of all infarcts and strokes occur before the age of 60
- Stary and colleagues established that for the first three decades or so of life, the disease gets a foothold in the artery, but it’s only in the fourth decade that one starts to develop the lesions that can actually precipitate a clinical event
- Risk is low, yet the event rate is high — How could that possibly be?
- Allan says the answer is “stunningly obvious” — Allan has published on how this can be explained; there are 2 problems:
- 1) There are a ton more people under 60 than over 60; so the rate of events is low, but the absolute number of events is high
- 2) Say a patient gets to their 60’s without a cardiac event, but the disease was developing and extending during their 30’s-50’s
- So by the time a doctor tries to prevent an event, the disease is well advanced in the arteries
- These are the 2 fatal flaws in the 10-year risk approach
- Published in JAMA Cardiology in 2018, A Long-term Benefit Approach vs Standard Risk-Based Approaches for Statin Eligibility in Primary Prevention
- Risk is a good concept but Allan realized doctors should be selecting people based on causes of CVD
- For example, if Peter’s risk is 4.1%, what does this number mean?
- Is his risk 4.1%? ⇒ No, it means 4.1 people out of 100 will have an infarct
- But within this category, there is tremendous variance in real risk
- Not everyone is at 4.1; some are higher; some are lower; some are dead on
- Is his risk 4.1%? ⇒ No, it means 4.1 people out of 100 will have an infarct
- The philosopher AJ Ayer (known for logical positivism) was actually darn good on probability
- There’s a real challenge predicting singular events
- One is either going to have an infarct in the next year or not; it’s not really a probability
- If one algorithm said a patient has a 10% risk and another one said a patient has a 15% or 20%, whether they have an infarct or not, both algorithms were right because they said there was sort of a chance the patient would have an infarct
- There is also a far greater chance that the patient won’t have an infarct
- When the advice is to treat patients with a risk abouve 7.5%, that means 92.5% of the time nothing will happen to these patients
- That’s not a great incentive for helping people understand what’s truly going to happen
A 30-year model of risk that focuses on cause of CVD
- What Allan has done is develop what’s called a casual benefit model
- Published in Clinical Science in 2012, The Causal Exposure Model of Vascular Disease
- Published in JAMA Cardiology in 2017, The Benefit Model for Prevention of Cardiovascular Disease: An Opportunity to Harmonize Guidelines
- With a measure of non-HDL or apoB, risk can be projected for over 20-30 years
- For a 30-year-old, the period of time they should care about is up to at least 60
- For example, this model could predict that a 35-year-old has a 30% chance of stroke before age 65
- This is a number that has meaning
- Now a second calculation can be made to determine how much the risk can be reduced by starting treatment at age 35 or how much the patient would lose by starting at age 45 or age 55
- When Allan gave Peter the book on atherosclerosis he was starting his own journey on trying to construct an alternative to the present risk model in which with the help of my colleague here at McGill, George Thanassoulis, and Michael Pencina from Duke, and Karol Pencina from Harvard we have done
- Published in JAMA Cardiology in 2017, The Benefit Model for Prevention of Cardiovascular Disease: An Opportunity to Harmonize Guidelines
- Peter remembers from pathology class in med school something the professor said, “No doctor has more experience with what it is to have heart attacks than pathologists because 50% of the people who have a heart attack die on their first heart attack.“
- So 50% of people who have a heart attack, their first presentation is death
- It’s a very sobering fact
- He doesn’t think this is true today, but it was 25 years ago
- Maybe now one third of first events are fatal; sobering nonetheless
- The textbook went through, in great detail, the pathological staging of atherosclerosis; it was also littered with autopsy sections of coronary arteries from people who had died for other reasons
- Notably, many of these people who died from other causes, but their coronary arteries were studied in autopsy, these people were quite young
- A 26-year-old male victim of a gunshot wound; or a 27-year-old female who died in a motor vehicle accident, etc.
- Looking at their coronary arteries, one realizes they already have atherosclerosis; they have oxidized apoB-bearing particles engulfed by macrophages and thickened intima
- While they may not have calcification in their arteries yet or the types of plaque that would rupture within the ensuing weeks or days or months, they nevertheless had atherosclerosis
- Now what Peter’s professor said some 20 years earlier made sense
- Notably, many of these people who died from other causes, but their coronary arteries were studied in autopsy, these people were quite young
- Allan remarks that cardiologists are so used to looking at angiograms
- “Oh, there’s a LAD lesion”
- A lesion in the left anterior descending artery
- “Oh, there’s a LAD lesion”
“Well, there’s an LAD lesion, but the whole damn artery is diseased. And when you destroy the normal architecture of the artery, you can’t restore it.” – Allan Sniderman
- Much of statin prevention therapy is to prevent complication of disease not actually prevent disease
- Statins lower apoB particle number; that’s how they work
- With fewer apoB particles in plasma, fewer get into the arterial wall, fewer get trapped
A primer on cholesterol, apoB, and plasma lipoproteins [16:30]
Cholesterol
- Cholesterol is a fat lipid
- It’s a critical element in cell structure; it’s in all cell membranes
- The amount that’s in the cell membrane is determinative of the function of the membrane
- All the cells in the body can synthesize cholesterol
- Only the liver can really break it down in any amount and excrete it
- When we eat, we absorb cholesterol and fatty acids in the form of triglycerides, and they get resynthesized in the intestine into particles
- Cholesterol and triglycerides cannot be transported because they don’t mix with water, they’re not soluble
- Triglycerides are fatty acids tacked onto a glycerol backbone; see the structure of a triglyceride in the figure below
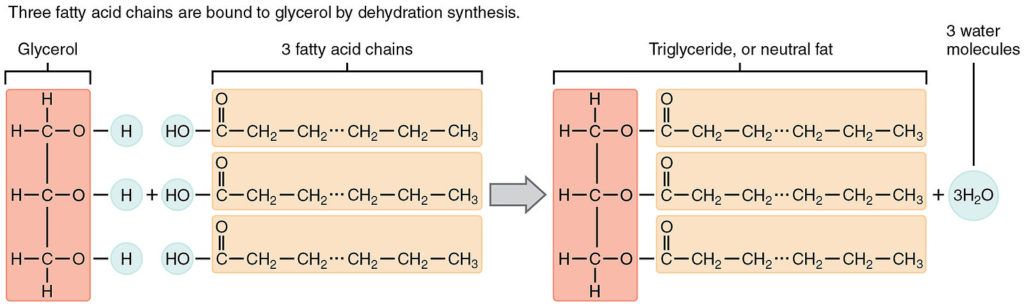
Figure 2. A triglyceride is made by attaching 3 fatty acids to a molecule of glycerol. Figure credit: Wikimedia Commons
{end of show notes preview}
Would you like access to extensive show notes and references for this podcast (and more)?
Check out this post to see an example of what the substantial show notes look like. Become a member today to get access.

Allan Sniderman, M.D.
Allan Sniderman obtained his MD from the University of Toronto in 1965 and then moved to Montreal, where he did his clinical training in Internal Medicine and Cardiology at McGill University. In 1971, he went to the University of California at San Diego to study lipoprotein metabolism with Dr. Daniel Steinberg. He returned to McGill and, with the passage of time, became the Edwards Professor of Cardiology and a Professor of Medicine at McGill University.
With colleagues within and outside McGill, he began and has continued a series of studies, which identified the commonest dyslipoproteinemia associated with coronary artery disease- hyperTg hyperapoB. Study of the pathophysiology of hyperTg hyperapB led to studies of the regulation of hepatic apoB secretion and the uptake and release of fatty acids by adipose tissue. He has conducted an extensive series of epidemiological studies, which have demonstrated apoB to be superior to LDL-cholesterol as a marker of the risk of vascular disease. His current research interests are: to understand the regulation of plasma LDL, to create simplified but advanced diagnostic algorithms to recognize and treat those with and those at high risk of vascular disease, and to develop new models to determine the absolute value of different strategies to identify and treat those at risk of vascular disease.
Dr. Sniderman, father of five, grandfather of four, is a committed, but not very skilful, golfer. His most joyful moments have come from learning with his students and colleagues.
Allan D. Sniderman, MD, is the Edwards Professor of Cardiology and Professor of Medicine at McGill University. He is Director of the Mike Rosenbloom Laboratory for Cardiovascular Research at Royal Victoria Hospital in Montreal and was elected a Fellow of the Royal Society of Canada in 2009. [McGill University Health Centre]



{kind=link}