In this “Ask Me Anything” (AMA) episode, Peter and Bob take a deep dive into fat flux. They define the major players that impact the flow of fat entering and exiting a fat cell, which determines how much fat a person carries. They discuss the significant influence that insulin has on the net fat balance and explore common strategies, such as fasting and low-carb diets, that have efficacy in the liberation and oxidation of fat from fat cells. Additionally, Bob explains his research process and how he seeks answers to Peter’s challenging questions.
If you’re not a subscriber and listening on a podcast player, you’ll only be able to hear a preview of the AMA. If you’re a subscriber, you can now listen to this full episode on your private RSS feed or on our website at the AMA #22 show notes page. If you are not a subscriber, you can learn more about the subscriber benefits here.

We discuss:
- The two main ways to reduce fat mass (1:30);
- Explaining fat flux—how fat enters and exits a fat cell (9:15);
- What fat balance looks like (21:15);
- What net fat influx looks like, and the impact of insulin in lipolysis (24:30);
- What net fat efflux looks like, and the benefits of fasting for breaking the hyperinsulinemic cycle (28:30);
- Exploring why most people with excess body fat will lose fat mass when reducing carbohydrates or eating a ketogenic diet (32:45);
- Why being in nutritional ketosis does not automatically translate to negative fat flux (fat loss) (42:40);
- Bob’s approach to scientific research (47:00);
- The importance of curiosity and a desire to learn (58:30);
- Bob’s tips and tricks for answering a scientific question in a time-crunch (1:00:00); and
- More.
Get Peter’s expertise in your inbox 100% free.
Sign up to receive An Introductory Guide to Longevity by Peter Attia, weekly longevity-focused articles, and new podcast announcements.The two main ways to reduce fat mass [1:30]
A lot of questions related to fat flux:
-Why doesn’t oxidation of fat necessarily mean you’re losing total body fat?
-If I eat a low carb diet and become a “fat burning machine,” why don’t I always lose fat on this diet?
-If I’m in ketosis, doesn’t it mean I’m burning fat?
-Peter’s response: “You can absolutely be in ketosis and gain weight. You can absolutely be a ‘fat burning machine,’ and still accumulate fat.”
Some shorthand to be used in today’s podcast:
- Using “gaining weight” and “gaining fat” interchangeably
- So when people say, “I want to lose weight,” what they really mean is “I want to lose fat.”
- When people say, “I want to gain weight,” they usually mean “I want to gain muscle.”
-Well, there are two main approaches to losing fat
- 1 – Reduce the total number of fat cells
- 2 – Shrink the fat cells
- The former (reduce the total number of fat cells) is most typically something that is done with liposuction
- Note: An NEJM study suggests that there is a profound difference between losing fat via liposuction vs. through dietary & lifestyle interventions
“We should never confuse the metabolic benefits that come from reducing the size of adipose tissue with reducing the amount of it. The former, in the case of liposuction, is really a cosmetic procedure. Whereas the latter of course has cosmetic benefits, but much more importantly as a metabolic improvement.” —Peter Attia
How do you shrink a fat cell? [6:12]
Overview:
- An engineer would think about this by drawing a boundary, looking at the boundary conditions, and effectively understanding what goes in and what comes out
- Mass cannot be created from nothing and mass cannot disintegrate into nothing
- If a fat cell gets larger, there is a net accumulation of fat in that cell relative to how much goes out of it (and the converse is true)
Quick example: A room has 100 people in it
- There were people traveling in and out of the room constantly so if you want to understand if that room is increasing or decreasing in the number of people in it, you need to understand what is happening at every point where there’s an entry or exit
- By doing that, you can understand what is the net increase or decrease
- Peter refers to that as flux
- So, today they will be looking at fat gain/loss through the lens of fat flux—i.e., What is the flux of fatty acids substrate into and out of a fat cell, and can we infer the behavior of that fat cell in response to that?
Bob adds:
- When we talk about weight loss or weight gain or fat loss or fat gain, a lot of times we’ll hear about calories in, calories out
- Basically, if any more energy is entering the system than leaving it, the system is getting bigger and vice versa.
- If more energy is leaving the system than entering it, it’s more or less getting smaller
- What Peter is talking about is at the level of the adipose tissue—i.e., how much fat is going into the adipose tissue, how much is being released, and also how much is being trapped
⇒ Check out Peter’s blog post: How to make a fat cell less not thin: the lessons of fat flux
Explaining fat flux—how fat enters and exits a fat cell [9:15]
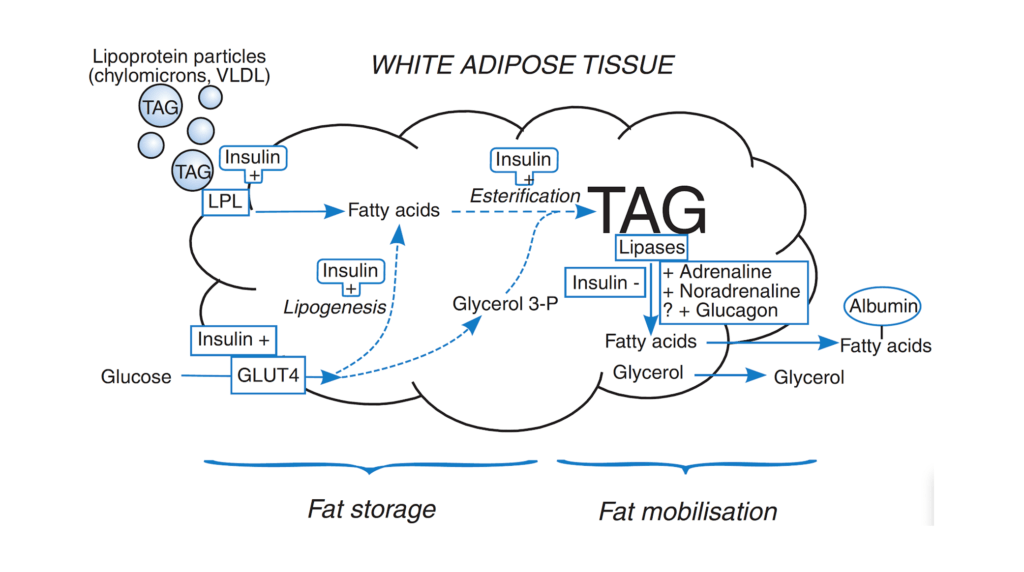
Figure 1. Overview of fatty acid and glucose metabolism in white adipose tissue. The body’s main store of chemical energy is in the form of TAG in WAT. Fat storage is the process of deposition of TAG; fat mobilization (or lipolysis) is the process of hydrolysis of the stored TAG to release NEFA into the plasma (bound to albumin), so that they can be taken up by other tissues. The major pathways and main sites of hormonal regulation are shown: a plus sign indicates stimulation, a minus sign inhibition. Dashed lines show multiple enzymatic steps. [Source: Frayn, 2010]
WAT vs BAT
- White adipose tissue (WAT) is called white adipose tissue to contrast it from brown
- BAT is a form of adipose tissue that is not as prevalent as WAT, but it has really unique metabolic properties, namely, a higher concentration of mitochondria and therefore a much greater metabolic activity
- We’re focusing on WAT, which is what most people think of when they think of fat — a glistening, yellow substance with cellular structure that looks surprisingly simple
- But white adipose tissue is an endocrine organ and is way more complicated than it looks under a microscope
Looking at the above figure, it has two sides:
- You have an “in” side
- and you have an “out” side
- On the left hand side of the above figure, there are 2 doors—the upper left door and the lower left door are two ways that the fat cell gets fatter
- On the right hand side of this page, you have the exit door—this is how the fat cell gets skinnier
The first door (upper left of figure):
- This is basically how we bring fat into a fat cell
- What you see in this figure are triacylglycerides and in this figure, TAG, which is synonymous with TG
- TAG means three fatty acids onto a glycerol backbone and that’s the way that we very efficiently store fat
- *IMPORTANT DISTINCTION: When you get your blood tests done… they’re looking at your triglycerides carried in your lipoprotein, most of which are VLDL cholesterol (they are NOT looking at TAGs in your fat cells)
- This test is nearly always done in a fasted state —
- if you get a score of 60, that’s good;
- if you get a score of 150, they doc will want you to lower that;
- That test is primarily measuring the amount of triglycerides in your VLDL cholesterol
- But if we measured your triglycerides as you were eating, we’d get a higher level for one and much of it would be carried in the chylomicron
- chylomicrons, which are very short-lived, are what bring fat that you eat in from the gut into the plasma, and then ultimately either get utilized or more commonly, get brought and stored here in the fat cell
- that’s why this picture is showing both the chylomicron, which is postprandially and the VLDL, which is most other times
- This test is nearly always done in a fasted state —
- Two things that are highlighted on the upper left side of the figure: i) LPL, and ii) insulin
- LPL stands for lipoprotein lipase (anything that ends in -ase is an enzyme)
- This lipase facilitates the transport of triacylglycerides into the fat cell
- Insulin:
- the reason that insulin is sitting there is insulin is the strongest regulator of that
- insulin activates and upregulates LPL
- Insulin is a very anabolic hormone—a building hormone—so it’s anabolic to muscle and unfortunately anabolic to a lot of cancer and to a fat cell
- insulin is pro growth, and it wants to bring more of these fats in
- LPL stands for lipoprotein lipase (anything that ends in -ase is an enzyme)
Second door (lower left side):
{end of show notes preview}
Would you like access to extensive show notes and references for this podcast (and more)?
Check out this post to see an example of what the substantial show notes look like. Become a member today to get access.



