
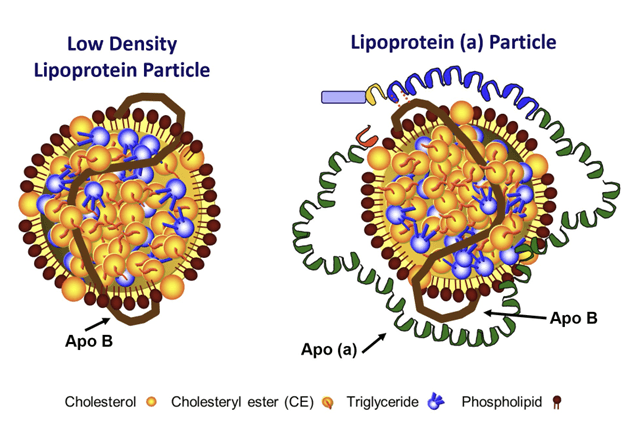
Since the approval of lovastatin in the U.S. 35 years ago, statins have been a game-changer in reducing low-density lipoprotein (LDL) levels and combating atherosclerotic cardiovascular disease (ASCVD). Broadly speaking, an LDL particle is a lipoprotein within a specific density range and is a collection of lipids (mostly cholesterol) enwrapped by a single protein called apolipoprotein B (apoB). But LDLs are actually a two-member family (see Figure below), as some LDL particles contain another surface protein called apolipoprotein (a), abbreviated apo(a), which binds to apoB. Such LDLs are what we call Lp(a) particles (pronounced “el-pee-little-A”), and elevated Lp(a) concentration is itself widely recognized as an independent risk factor for ASCVD. (In the rest of this discussion, LDLs with apoB but no apo(a) are referred to as “real LDL” particles and those having both apo(a) and apoB are referred to as Lp(a) particles.)
Despite the enormous benefit of statin medications in reducing LDL, ongoing concern lingers over the observation that, in certain patients with coronary artery disease (CAD), statin therapy results in elevated concentration of Lp(a). But recently, during a presentation at the Trans Catheter Cardiovascular Therapeutics meeting in Boston, Minatoguchi et al. of the Mount Sinai Health System in New York presented new data on the effect of high-intensity statins on both Lp(a) and coronary lesions. Their results, which will now be subject to peer review and the publication process, provide food for thought and generate a reassuring hypothesis regarding statin-induced Lp(a) elevation.
Motivation for the Study
CAD is characterized by the buildup of plaques in the arteries which serve as the heart’s blood supply. These plaques are composed of a lipid-rich core primarily made up of oxidized lipids, including cholesterol and cholesterol crystals. Overlying the lipid core is a fibrous cap generated by smooth muscle cells, and the thickness of this cap determines the potential for plaque rupture and clot (thrombus) formation, a main cause of heart attacks and strokes. The thicker the cap, the less likely the plaque is to rupture.
Some (but not all) previous work has shown that elevated Lp(a) is associated with increased incidence of coronary events in patients with high levels of LDL cholesterol (LDL-C) but not necessarily in patients with low LDL-C (at least in unmedicated patients). Statins, which enhance clearance of real LDLs by increasing levels of the LDL receptor (LDLR) in the liver, are not effective at likewise increasing clearance of Lp(a), as apo(a) prevents the apoB on LDLs from binding to LDLR. Not only are statins ineffective at reducing Lp(a), they may, in some people, even increase Lp(a) particle production by inducing the synthesis of apo(a). Thus, we see the rationale for Minatoguchi et al’s study: if statins raise Lp(a) while lowering the burden of real LDL, what is the overall effect on plaque progression and ASCVD risk? After all, this is the only thing that matters.
About the Study
The authors used intravascular ultrasound and optical coherence tomography to determine how statin-induced changes in Lp(a) affected plaque progression and quality. A total of 85 participants (mean age of 62 ± 11, 58% male) on high-dose statin therapy (rosuvastatin 40 mg) were divided into two groups – low and high – depending on baseline Lp(a) levels. In the “low” and “high” groups, mean Lp(a) concentrations were 44.5 ± 51.6 mg/dL and 118.8 ± 50.3 mg/dL, respectively, with the high Lp(a) group also demonstrating significantly thinner (84 ± 32 vs. 103 ± 44 µm, p=0.046) minimal fibrous cap thickness (FCT) – the thickness of the thinnest (and thus weakest) point in the cap. The duration of treatment was not stated in the author’s brief presentation but is expected to be included in their upcoming publication of this work. (We assume a period of at least 4 months based on previous work indicating that this is the length of time it takes for statins to increase Lp(a).)
After statin treatment, Lp(a) in the low group rose significantly to 60.4 ± 70.4 mg/dL (p<0.001), leading to an additional 6 patients in the high Lp(a) group. The change in the high group was not reported but is expected to be comparable to the low group in relative percent increase. Yet despite the increase in Lp(a), FCT had improved in both groups at follow-up, though changes were not significant (103.0 vs. 107.2 µm for the low group, p=0.64 and 84 µm vs. 103 µm for the high group). The lack of effect on – or even improvement in – FCT led the authors to conclude that the increase in Lp(a) on high-intensity statin therapy was not an independent predictor of changes in FCT and does not appear to be of clinical significance.
Making Sense of the Results
As I’ve discussed on the podcast with Dr. Benoît Arsenault, Lp(a) is the single most important genetically-inherited trait driving ASCVD risk, so the question remains: why isn’t the Lp(a) increase clinically significant?
Total LDL particle concentration (LDL-P) is the sum of real LDLs + Lp(a). Likewise, LDL-C represents the cholesterol within all of the circulating LDLs (real LDLs + Lp(a) particles). Particle for particle, Lp(a) particles are more atherogenic than real LDLs. However, over 90% of all LDL particles in circulation are of the latter, apo(a)-negative group, and in the majority of people, Lp(a) particle number is quite small and thus does not contribute to total LDL-P or total LDL-C. Even among the 20% of people who have inherited the ability to produce high enough numbers of Lp(a) particles that it contributes significantly to their ASCVD risk, Lp(a) is still a minority LDL particle. Whether elevated or not, Lp(a)-P levels never approach those of real LDLs.
Statins are only effective at clearing real LDL and not Lp(a) (again, due to apo(a) interfering with particle binding to the LDLR), but Lp(a) accounts for only a small percentage of total LDL. So despite the fact that these medications do not reduce concentrations of Lp(a) – and may even increase it by 5-20% – they are so effective at clearing real LDLs from circulation that total LDL-P or apoB is significantly reduced despite any Lp(a)-P increase. In other words, the effect of statins on real LDLs – which constitute the overwhelming majority of apoB particles – is so profound that it dominates the net impact of these drugs on ASCVD risk.
The Bottom Line
All current Lp(a) guidelines, fully cognizant of statins’ potential ability to increase Lp(a), suggest that a mainstay of reducing Lp(a)-related ASCVD risk is to consider statin therapy. So although trials have long established that statins can increase Lp(a) concentrations, the Minatoguchi et al. study lends support to the idea that such increases are not of clinical importance. Any change in risk associated with an increase in Lp(a) is overwhelmed by the magnitude of the effect of statins on real LDLs; and therefore, no one should cease statin therapy due to any increase in Lp(a).
For a list of all previous weekly emails, click here.


