
Pharmaceuticals are one of modern medicine’s tools to prevent and treat acute and chronic diseases – everything from treating cancer to lowering plasma lipids for the prevention of atherosclerotic cardiovascular disease (ASCVD). Yet the process of drug discovery and development is long and expensive, and the vast majority of drugs that enter this pipeline will ultimately fail to reach the point of FDA approval. The expense and high failure rate can contribute to the high price tags on many medications that do end up on the market (as discussed in depth by Saum Sutaria on a recent episode of The Drive). But why do so many drug candidates fail? And how might the drug development process be improved in order to better allocate resources toward the most viable options?
Stages of drug development
Before we can evaluate potential areas for improvement, we first need to understand some basics of the current process of drug development.
In the earliest stages, candidate drugs undergo years of testing and development to evaluate efficacy and potential toxicity through in vitro cell culture and in vivo animal studies. Preclinical animal studies are also used to determine initial human dosing, scaled by relative differences in weight.
Before testing can proceed from animals to humans, the drug’s sponsor (often the manufacturer) must then submit an investigational new drug (IND) application. This application includes the preclinical efficacy and toxicology data, detailed protocols for the initial clinical trial phases, and manufacturing information meeting the requirements for “Good Manufacturing Practices” (GMP) – criteria that establish, for instance, a manufacturer’s ability to consistently supply a drug for clinical trials with the necessary quality controls. The FDA reviews IND applications to gauge the risks to research study participants and rejects the application if risk is deemed too high or if there are study design flaws. Once an IND application is approved, the clinical trial process can begin.
The table below delineates the four or five clinical research study phases, each of which builds upon the last – if the drug moves on to the next phase.
Table: Phases of Clinical Research

The most recently introduced phase 0 is not mandatory but is intended to expedite the clinical evaluation of new drugs, for instance by demonstrating that a given drug reaches its intended target tissue. (If a new cancer therapy does not reach a tumor, there is little point in proceeding to studies in a larger population.)
The four subsequent phases test drug safety and efficacy, determine dosage, and compare the new drug to placebo or other standard treatments. Early in the process, the focus is primarily on safety, with efficacy and comparison to alternatives taking on greater importance in later phases. Thus, when I occasionally highlight new drugs that are showing promise in early stages (I or II), any results are just that – promising, but not guaranteed to hold up in a more stringent randomized controlled trial (RCT) in phase III.
Phase III is the first phase of clinical drug testing that requires comparison to a placebo group – or in cases where it would be unethical to give no intervention, a comparison to the existing standard of care. Some more recent drug trials use placebo comparisons in phase I, Ib, and II testing (for instance, this was the case with the triple receptor agonist retatrutide), though this practice is not required at these stages.1–4 Just as phase 0 is intended to expedite the clinical evaluation of a drug, it may become more commonplace to have a placebo or other comparator groups in earlier phases to preview the drug’s expected performance in the most important phase III trial. Earlier trial phases take years and cost tens of millions of dollars to conduct, but phase III is the one that really matters. Successful phase III trials provide the safety and efficacy data necessary for a drug to then receive approval by regulatory agencies for subsequent commercial sale. After a drug hits the market, data on efficacy and safety continue to be monitored (known as “phase IV” testing) across the populations of individuals using the medication.
Where do drugs typically fail?
Most drug candidates – roughly 90% – fail at some point between preclinical studies and the conclusion of a phase III trial. The figure below is a broad-stroke visual representation of the estimated success at each phase of drug development, as well as the relative cost as of 2003,5 though more recent estimates indicate that the success rate may be even lower.6 Additionally, note that costs are much higher today than the numbers reported 22 years ago. (To bring a new drug to market costs on average about a billion dollars, so each of these trial phases likely costs about 8-10x more than in 2003.) This figure also does not include failures that occur after a drug receives regulatory approval, as post-market surveillance occasionally reveals previously undetected safety concerns which can lead to a drug’s withdrawal. Though noteworthy, these occurrences are fairly rare – on average, approximately one drug has been withdrawn each year in the US over the last four decades due to post-market safety issues (compared to roughly 30-60 drugs which are typically approved by the FDA each year).6,7
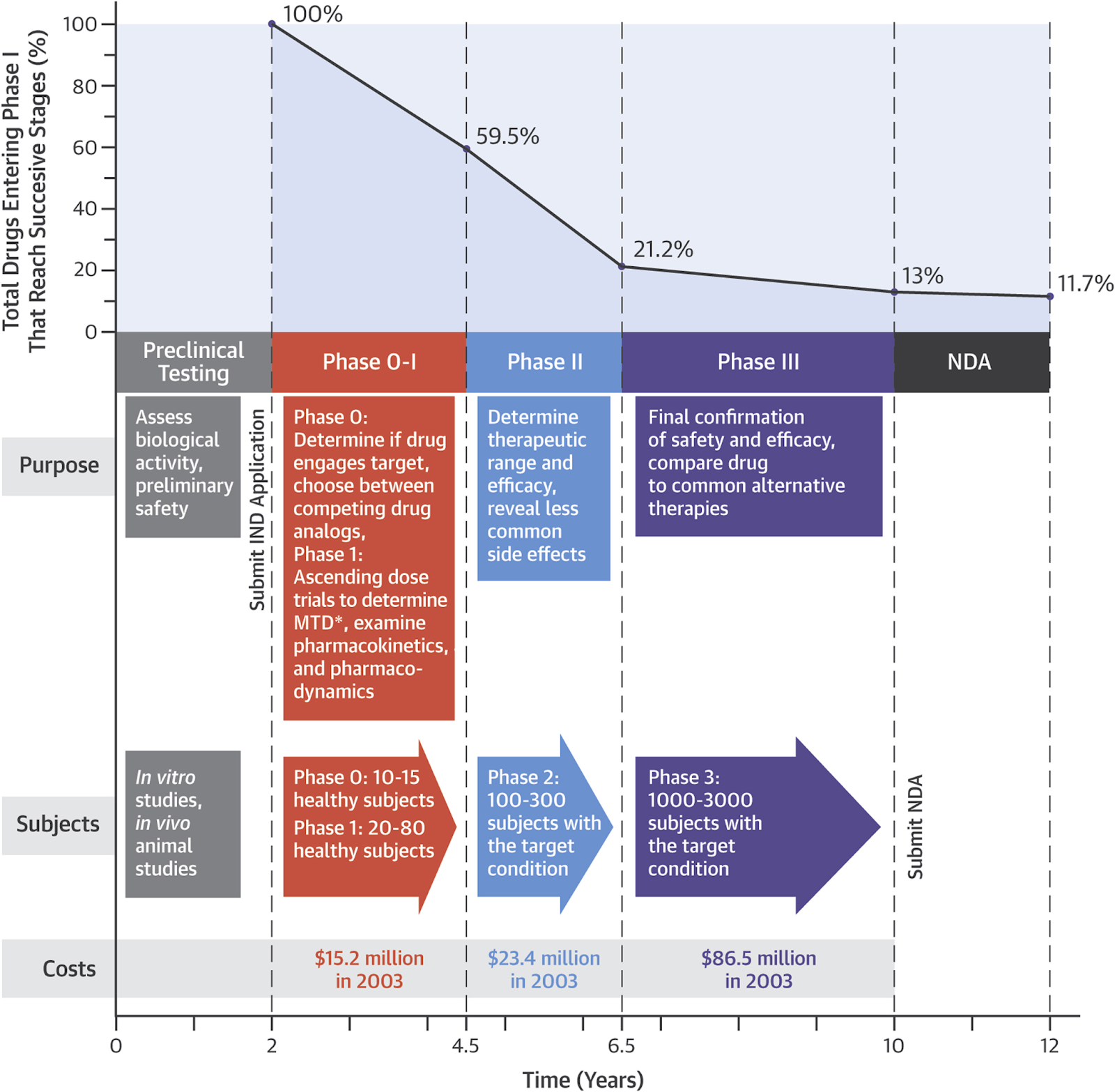
In all, drugs failing to show an expected therapeutic effect account for the largest percentage of failures in drug development – between 40-50%.8 Biological discrepancies between cultured cell lines, animal models, and humans make it difficult to validate if a drug is successfully reaching and affecting its intended molecular target. Further, in order for a drug to receive approval, the FDA must deem efficacy to be not only statistically significant, but clinically significant as well. Study outcomes are usually set before the initiation of a phase III trial by the drug company, not the FDA or a third party, and a “successful” study outcome doesn’t automatically mean an outcome with true clinical value. This is also why approved uses are limited to the studied population. An antidiabetic drug that improves glycemic control and induces significant weight loss, isn’t approved for on-label use in treating obesity until an obesity-specific trial is run.
Another 30% of failures are attributed to toxicity, either due to off-target effects or the accumulation of the drug in human tissues. Potential risks are weighed against benefits, taking into account the severity and likelihood of a given adverse event occurring, as well as the intended use of the medication, which can shift the balance point of risk to reward. For instance, the level of risk that might be considered acceptable would likely be much higher for a new drug to treat pancreatic cancer than it would be for new drugs to treat headaches or blood pressure.
How might we improve?
Failures are bound to be a common occurrence in any rigorous system for drug development. But various strategies offer realistic opportunities for improvement in rates of success, as well as in the speed with which we might identify likely failures. By abandoning doomed drugs earlier in the process, vast amounts of time and money might be saved and redirected toward more promising candidates.
One proposed improvement involves using a two-parallel-arm approach in the preclinical phase. The most common preclinical studies evaluate drugs’ efficacy and toxicity by their “structure-activity relationship” (SAR), which helps to determine which portion of the drug is responsible for the effect. But the addition of an evaluation of the “structure‒tissue exposure/selectivity relationship” (STR)9 allows further assessment of the accumulation of a drug in its target tissue and in normal, healthy tissues. A drug’s plasma exposure is not necessarily correlated with efficacy, so if a drug is optimized for its molecular target but does not reach the desired tissue, this STR analysis would signal a need to modify the drug structure in order to have the desired therapeutic effects. When testing multiple drug structures, the results of these two study arms create a hierarchy of efficacy and toxicity that potentially improves clinical translation.
In later stages of drug development, more widespread use of adaptive trial designs may also help to maintain rigor of the testing process while cutting costs. In such designs, the study protocol is permitted to change as new information – such as preliminary study results – come to light.
However, the most impactful advancements will likely arise from the use of artificial intelligence (AI) and machine learning, which are already substantially accelerating all stages of the drug development process. AI algorithms can sift through large datasets (e.g., “omics” data) to identify novel drug targets or genes and even aid in drug design by predicting the three-dimensional structure of the target molecule. Once a target is identified, AI can screen large libraries of molecules for potential drug candidates as well as generate de novo drug designs. Such an approach might also identify new potential indications for existing drugs, which, as discussed in a recent newsletter, can drastically accelerate the path from concept to approval. Further, AI simulations can be used to predict drug activity and safety, reducing the number of necessary animal studies. The widespread use of AI in drug development has the potential to both lower the cost of bringing a drug to market and reduce the development time. The lower costs of development and AI-enabled drug discovery will potentially incentivize companies to devote more time and resources to developing therapeutics for rare diseases since currently, the high cost of development outweighs any return on investment from a limited market.
The bottom line
The drug development process has advanced considerably over the last century to enhance rigor. Yet the process is also extraordinarily costly and time-consuming, with plenty of room for improvement with better study designs and the integration of AI. These efforts, in conjunction with changes in business practices on the part of drug companies (as detailed by Dr. Sutaria), may help to curb rising drug prices as well as to incentivize greater attention toward the development of therapies for otherwise ignored rare diseases.
Some percentage of failures, even relatively late in the clinical trial pipeline, are unavoidable. Translatability between animals and humans – and even variability in drug metabolism and efficacy across individual humans due to factors such as age, genetics, or body composition – are difficult to predict. This may change with further advancements in AI and computing, but until AI can reliably predict the risks and benefits right down to the level of the individual, phase III (and IV) clinical testing in large, diverse populations are still bound to uncover flaws that may frequently lead to the abandonment of a given drug prior to approval. But even as we seek to reduce the percentage of such failures, we must keep in mind that their very existence is proof that the process is doing exactly what it is meant to do – ensuring a high degree of reliability in the efficacy and safety of approved medications.
For a list of all previous weekly emails, click here.
References
1. Coskun T, Urva S, Roell WC, et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab. 2022;34(9):1234-1247.e9. doi:10.1016/j.cmet.2022.07.013
2. Urva S, Coskun T, Loh MT, et al. LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: a phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial. Lancet. 2022;400(10366):1869-1881. doi:10.1016/S0140-6736(22)02033-5
3. Jastreboff AM, Kaplan LM, Frías JP, et al. Triple-hormone-receptor agonist retatrutide for obesity – A phase 2 trial. N Engl J Med. 2023;389(6):514-526. doi:10.1056/NEJMoa2301972
4. Rosenstock J, Frias J, Jastreboff AM, et al. Retatrutide, a GIP, GLP-1 and glucagon receptor agonist, for people with type 2 diabetes: a randomised, double-blind, placebo and active-controlled, parallel-group, phase 2 trial conducted in the USA. Lancet. 2023;402(10401):529-544. doi:10.1016/S0140-6736(23)01053-X
5. Van Norman GA. Drugs, devices, and the FDA: Part 1: An overview of approval processes for drugs. JACC Basic Transl Sci. 2016;1(3):170-179. doi:10.1016/j.jacbts.2016.03.002
6. Dowden H, Munro J. Trends in clinical success rates and therapeutic focus. Nat Rev Drug Discov. 2019;18(7):495-496. doi:10.1038/d41573-019-00074-z
7. Rodriguez-Monguio R, Seoane-Vazquez E, Powers JH 3rd. A comparative assessment of approvals and discontinuations of systemic antibiotics and other therapeutic areas. Healthcare (Basel). 2023;11(12). doi:10.3390/healthcare11121759
8. Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022;12(7):3049-3062. doi:10.1016/j.apsb.2022.02.0029.
9. Gao W, Hu H, Dai L, et al. Structure‒tissue exposure/selectivity relationship (STR) correlates with clinical efficacy/safety. Acta Pharm Sin B. 2022;12(5):2462-2478. doi:10.1016/j.apsb.2022.02.015



