There is strong scientific consensus that high levels of certain cholesterol-trafficking particles, such as low-density lipoprotein cholesterol (LDL-C), play an important role in the development of atherosclerotic cardiovascular disease (ASCVD). Yet this foundational understanding of the pathogenesis of heart disease has recently been challenged by a documentary called The Cholesterol Code, igniting fierce debate on social media about what we know about cardiovascular health. The film, associated with an organization called the Citizen Science Foundation (CSF) that conducts independent research funded by private donations, argues that even very high LDL cholesterol levels might not always indicate ASCVD risk. At the center of their controversial hypothesis is a subset of individuals they have termed “lean mass hyper-responders” (LMHRs): people who develop remarkably high LDL-C levels after adopting a low-carbohydrate, high-fat, ketogenic diet, yet maintain a specific cluster of other favorable metabolic markers. Whether such a specific phenotype exists is itself open to debate, but the truly controversial part of the hypothesis is the suggestion that serum LDL-C may not drive ASCVD in LMHRs. More than an academic exercise, this debate cuts to the core of how we think about cardiovascular risk and challenges decades of painstaking scientific work. Before getting into the details, it’s worth stating the central issue plainly: Atherosclerosis is driven by the number of apoB-containing particles in circulation, and that relationship holds regardless of how or why those particles are elevated. The question is not whether LMHRs are metabolically distinct, but whether they are exempt from this biology.
***
Two quick thoughts before we start:
First, we recognize that some of the discussion ahead will be dense with biochemistry. There is simply no way around that. Understanding the arguments put forth by the CSF, and by extension our counterarguments, requires going beyond sound bites and social media discourse. We have worked to strip away unnecessary verbiage while preserving the depth and rigor of the content. We encourage everyone, whether you agree, disagree, are undecided, or are simply confused, to engage with this material thoughtfully.
Second, decades of misleading guidance from health authorities, worsened by the growing entanglement of science, which is a process of discovery, and advocacy, which serves an agenda, have fueled widespread distrust in scientific institutions, particularly when it comes to human health. In many cases, this skepticism is understandable and even justified. However, while it would be naive to accept every medical dogma at face value, an even greater mistake, and one with serious consequences, is to reject scientific consensus entirely.
Ok, let’s dive in.
Understanding lipids and lipoproteins
Cholesterol is an essential molecule for every cell in the body. It serves as a crucial component of cell membranes and as a precursor for bile acids and various hormones. The liver is the primary producer of the cholesterol distributed in plasma, while the brain produces its own separate supply. Dietary sources contribute a much smaller fraction. Cholesterol produced by the liver must then be trafficked to other parts of the body, but this task is complicated by a fundamental challenge: Cholesterol cannot travel freely through the bloodstream because it is not water-soluble.
This is where lipoproteins come into play. Think of lipoproteins as specialized vehicles, submarines if you will, that transport fat-soluble molecules through the bloodstream by encasing them in a hydrophilic, water-soluble shell composed of proteins and phospholipids. In addition to cholesterol, the major molecules transported by lipoproteins include triglycerides, that is, fats, which are the body’s primary form of stored energy.
Lipoprotein particles come in different sizes and densities, ranging from large, buoyant (thus very low density), lipid-rich chylomicrons to small, lipid-poor high-density lipoproteins (HDLs), as shown in Figure 1. Various lipoprotein classes serve different roles and some may beget others. For instance, some very low-density lipoproteins (VLDLs) gradually convert into low-density lipoproteins (LDLs) in the bloodstream as core triglycerides and surface phospholipids are enzymatically removed for use by the body.
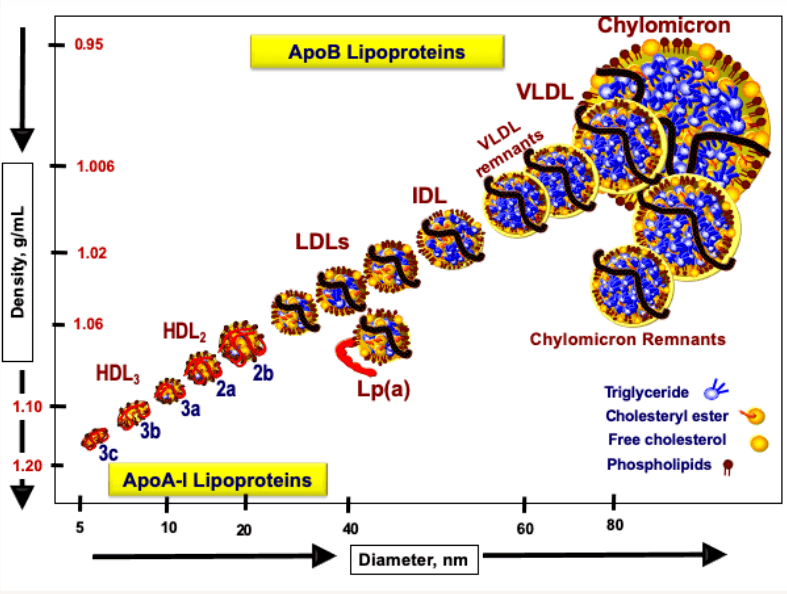
Crucially, however, only a few lipoprotein classes are capable of entering artery walls and initiating atherosclerosis. Specifically, atherogenic lipoproteins are those that are wrapped by a single molecule of a protein called apolipoprotein B (apoB). ApoB-containing lipoproteins include IDLs, LDLs, VLDLs, chylomicrons, and chylomicron remnants, but because LDL particles have a much longer plasma residence time than other apoB-containing lipoproteins, the vast majority, roughly 90 to 95 percent, of the circulating apoB pool is attributable to LDLs. Because the apoB pool is dominated by LDLs, and because each LDL particle contains only one molecule of apoB, plasma apoB level essentially serves as a measure of the number of LDL particles, which in turn closely approximates the total number of atherogenic lipoproteins in circulation.
(The older way to estimate the number of LDL particles was to measure the mass of cholesterol collectively carried by those particles, that is, “LDL cholesterol,” based on the assumption that an average LDL particle contains a predictable amount of cholesterol. The problem with this approach is that the relationship between LDL-C and LDL particle number is skewed in a significant fraction of the population, such that LDL-C can under- or overestimate true risk. But now we can measure apoB directly, giving us a more accurate way to gauge ASCVD risk than LDL-C alone.2)
Our research team spends hundreds of hours each month vetting studies and distilling dense literature to deliver evidence-informed insights on health and longevity. If you find value in our work, consider becoming a premium member and supporting our mission.











The LMHR hypothesis
So where does the concept of “lean mass hyper-responders” fit in? The CSF has identified what it believes to be a specific phenotype: individuals who, after adopting a low-carbohydrate or ketogenic diet, exhibit a distinct metabolic response marked by a characteristic triad of lipid markers. First, their LDL-C rises dramatically, often exceeding 200 mg/dL and sometimes reaching levels above 400 mg/dL. For comparison, the American Heart Association recommends keeping LDL-C below 100 mg/dL, or below 55 mg/dL for those at high risk of ASCVD.3 This rise in LDL-C corresponds to increases in plasma apoB concentration.4,5 The second feature in the triad is that HDL cholesterol (HDL-C) rises to unusually high levels, typically above 80 mg/dL. Third, triglycerides remain relatively low, usually below 70 mg/dL. This lipid phenotype is particularly striking because it differs from traditional patterns of dyslipidemia, in which elevated LDL-C often accompanies elevated triglycerides and relatively low HDL-C.
The CSF further argues that these individuals share several characteristics beyond their lipid profiles. They tend to be lean, typically with body fat percentages below 15 percent for men and below 25 percent for women, physically active, and metabolically healthy by conventional markers such as fasting glucose, insulin sensitivity, and inflammatory markers. Many are endurance athletes or engage in regular strength training.
According to the CSF’s hypothesis, this phenotype represents a distinct metabolic adaptation to low-carbohydrate, high-fat nutrition rather than a pathological state. They note that such sharp increases in LDL-C on a ketogenic diet are observed only in lean individuals with BMI below 25 kg/m2, not in those classified as overweight or obese,6 and they propose that these individuals are particularly efficient at mobilizing and utilizing fat for energy, giving rise to their unique lipid profile. In this view, elevated LDL-C is proposed to be a marker of enhanced fat metabolism that may carry a distinct cardiovascular risk profile. Some proponents go further and extrapolate from this to suggest that high LDL-C can therefore be ignored.
The “lipid energy model”
To explain this possible LMHR phenomenon, the CSF has developed what it calls the “lipid energy model,” which proposes a complex cascade of metabolic adaptations in response to carbohydrate restriction (Figure 2).7
When LMHR individuals restrict carbohydrates, their liver glycogen stores, the stored form of glucose, become depleted, triggering increased reliance on fatty acids rather than glucose for fuel. This shift supposedly leads to decreased basal insulin and leptin levels, which in turn increase lipolysis in fat cells, the process by which triglycerides are broken down into non-esterified fatty acids (NEFAs) for release into the bloodstream. With more NEFAs in circulation, more are taken up by the liver, where they are repackaged into triglycerides. As hepatic triglyceride synthesis rises, secretion of triglyceride-rich VLDL particles also supposedly rises. These VLDLs are then rapidly metabolized in the periphery, with triglycerides removed through the action of an enzyme known as lipoprotein lipase. As triglycerides are removed from VLDLs, the particles become LDLs, ultimately resulting in elevated LDL-C levels, or so the model posits.7
The CSF argues that this process is fundamentally different from other conditions that cause elevated LDL-C, such as familial hypercholesterolemia. They suggest that in LMHRs, high LDL-C reflects a higher-turnover state in which lipoproteins are delivering more energy to tissues and may therefore accumulate in arterial walls to a lesser extent than in other conditions with similarly elevated LDL-C. They further propose that the consistently low triglycerides and high HDL-C seen in these individuals indicate efficient lipid metabolism and protective adaptations.
However, as we will examine in detail, there are significant problems both with the model itself and with the evidence used to support it. While the observation of this lipid pattern in certain low-carb dieters appears to be real, the interpretation of its significance and the mechanistic explanations proposed to account for it face serious scientific challenges.
Examining the evidence
Much of the alleged support for the LMHR hypothesis comes from a pair of related studies highlighted in The Cholesterol Code documentary.
The primary study is a one-year prospective observational cohort focused on the development of atherosclerosis in LMHRs.5,8 The study aimed to monitor the development and progression of coronary atherosclerosis in 100 LMHR or “near-LMHR” individuals on ketogenic diets using coronary computed tomography angiography (CCTA) scans. Subjects were required to have maintained a ketogenic diet for a minimum of two years prior to the study start and to exhibit elevated LDL-C, at least 190 mg/dL, with the median among participants around 260 mg/dL, as well as relatively high HDL-C, at least 60 mg/dL.
The investigators reported that over one year, plaque progression (median noncalcified plaque volume progression of 5.6 mm³) was not associated with baseline apoB, change in apoB, or total LDL-C exposure, whereas baseline plaque burden was associated with subsequent plaque progression. In other words, the headline result was essentially that plaque begets plaque, while apoB and LDL-C did not predict short-term plaque progression in this cohort.
Even taken at face value, these results are not informative for the question being asked, because they examine the wrong timescale. Atherosclerotic risk is a function of cumulative exposure to apoB-containing lipoproteins, often conceptualized as the integral of apoB concentration over time. A one-year observational window in a cohort with decades of prior exposure heterogeneity is therefore structurally incapable of detecting the effect it is attempting to measure. The relevant question is not whether apoB predicts plaque change over a single year, but whether sustained exposure over many years increases cumulative disease burden. By design, this study cannot answer that question.
A more informative design would have included a control group of individuals with similarly high LDL-C but a more conventional dyslipidemic profile, such as low HDL-C and high triglycerides. If one year were truly enough time to assess whether high LDL-C drives ASCVD progression in LMHRs, then an appropriate non-LMHR, high-LDL-C control group should show measurably greater progression over that same period than would be predicted based on their additional risk factors alone. But the investigators included no such comparator. That omission makes it difficult to interpret the study as evidence that the LMHR phenotype is meaningfully distinct from more conventional forms of severe LDL-C elevation. Even this design would have remained imperfect, since such groups would still differ in important ways beyond their lipid profile, including blood pressure and insulin resistance, both of which influence ASCVD risk.
Although it might have taken longer to demonstrate results, an even stronger control group would have been individuals with low or moderate LDL-C but whose other risk factors were aligned with those of the LMHRs. If the risk associated with elevated LDL-C is truly abrogated in LMHRs, they should have shown no more plaque progression than this healthy control group. Again, however, no such control group was recruited.
To partly address this limitation, the authors point to a prior cross-sectional study published in 2024 that did include matched controls.9 This study compared 80 LMHR individuals who had been following a ketogenic diet for an average of 4.7 years with controls from the Miami Heart cohort study matched for age, gender, race, hyperlipidemia, diabetes, hypertension, and smoking status. The groups differed dramatically in LDL-C levels, with a mean LDL-C of 272 mg/dL among the LMHR group (here called the “KETO” group), with some values reaching 591 mg/dL, compared to a mean of 123 mg/dL among controls. The study used CCTA data to compare coronary plaque between groups and to correlate LDL-C with plaque levels. Results showed no differences in coronary plaque burden between the two groups and no correlation between LDL-C level and CCTA coronary plaque. But the question remains: Does elevated LDL-C in LMHRs pose less risk than a traditional dyslipidemic profile?
As noted above, analysis of the primary study alone is enough to raise major doubts about the results, because the most obvious control group was omitted and the study duration was simply too short to show progression of atherosclerosis from baseline. But when we turn to the companion study, several additional flaws become apparent.
First, this study was strictly cross-sectional, meaning data were collected at a single time point for a one-time comparison between groups. No metrics were tracked over time, which would have provided much stronger insight into how a ketogenic diet affects LDL-C and how those elevations then relate to ASCVD development. Atherosclerosis is the result of the cumulative exposure of a person’s arteries to apoB-containing lipoproteins over years and decades, not where their lipids are on a single given day. People in the LMHR group were on average 55.5 years old, and per protocol, they had had relatively normal LDL-C levels (122 ± 36 mg/dL) prior to adopting a ketogenic diet, and tested negative for genetic familial hypercholesterolemia. This implies that prior to their 4.7 years of diet-induced hypercholesterolemia, people in the KETO group had up to five decades of moderate apoB exposure. Conversely, the equally-high LDL-C in the control subjects in Miami Heart at the moment in time when they were matched with the KETO group likely reflects decades of exposure to such high levels. These patients’ apoB at the moment of the cross-sectional study will fail to capture their very different history of exposure, which likely explains much of the worse atherosclerosis in the Miami Heart controls relative to the KETO group.
Second, the matching process was inadequate. While the study matched for traditional risk factors such as age, gender, race, and smoking status, it failed to account for other crucial variables that could influence cardiovascular risk. People who voluntarily maintain a strict ketogenic diet for nearly five years likely differ significantly from the general population in physical activity, socioeconomic status, overall diet quality, and many other lifestyle factors (i.e., healthy user bias). None of these variables were matched or controlled for, yet any one of them could have influenced the results. In addition, the KETO group showed significant baseline advantages beyond their lipid profile. Compared to participants in the Miami Heart study, they had lower BMIs (22.5 vs. 25.8), lower inflammation markers (hsCRP: 0.5 vs. 0.7 mg/L; P = 0.007), higher HDL-C, and lower triglycerides. While these differences may be part of the so-called LMHR phenotype, they nevertheless make it impossible to isolate the specific effect of elevated LDL-C on cardiovascular risk.
Thus, rather than showing that LMHRs have a special response to ketogenic diets that somehow renders high LDL-C harmless, the much simpler interpretation is that the cardiovascular risk attributable to high LDL-C is consistent across individuals, but that the Miami Heart cohort’s ASCVD risk was elevated to levels similar to the LMHRs by longer exposure to elevated apoB, poorer metabolic health, higher inflammation, and likely inferior physical activity, diet, and socioeconomic status. In other words, the LMHR group was simply healthier than the control group in countless ways other than their LDL-C at the moment of the cross-sectional study, and those advantages collectively blunted the negative effect of their higher lipid levels.
Scrutinizing the mechanistic model
Despite the problems with the data used to support the LMHR hypothesis, is the “lipid energy model” still theoretically sound? On closer inspection, even this mechanistic foundation begins to crumble.
Any viable model must explain the magnitude of apoB elevation. The lipid energy model fails on all three fronts: it does not account for the observed increase in particle number, it contradicts the low triglyceride levels that define the phenotype, and it lacks direct experimental validation of its key steps.
As described earlier, the core of the model is that increased lipolysis leads to increased hepatic triglyceride synthesis and increased secretion and turnover of VLDLs, ultimately resulting in elevated LDL-C levels. However, this explanation has several fundamental problems. For one, the degree to which the liver increases triglyceride synthesis in response to lipolysis in fat cells is unclear, since NEFAs released from fat cells are distributed to tissues within minutes, and it is unknown how much actually reaches the liver in quantities sufficient to stimulate meaningful increases in triglyceride synthesis and secretion. More importantly, when the liver produces more triglycerides, it primarily increases the size of VLDL particles by adding more triglyceride cargo, but it does not substantially increase the number of VLDL particles secreted. This means that increased secretion of triglycerides does not significantly increase the total plasma concentration of apoB-containing lipoproteins, the crucial variable driving atherosclerosis, and therefore cannot account for the enormous elevation in apoB observed in alleged LMHRs.10
Furthermore, even if there were a large increase in VLDL particle numbers, we would expect to see elevated plasma triglycerides, since triglycerides within VLDLs are the primary contributors to total serum triglyceride concentration. But recall that one of the hallmark features of LMHRs is low triglyceride levels. Some supporters of the model argue that high HDL-C levels in these patients indicate high triglyceride turnover and thus explain the low triglycerides, but at present there is no evidence that this is the case—and if it were, it wouldn’t solve the flaw in the model, because high VLDL particle numbers would still lead to elevated steady-state levels of serum triglycerides. This explanation feels a bit like moving the goalposts, where each rebuttal to a central claim is met with a new, untested explanation. A more likely explanation is that high HDL-C levels are caused by HDL particles accepting more effluxed cholesterol as a result of increased cellular cholesterol synthesis, a known consequence of increased saturated fat intake.
Finally, the model proposes that high LDL concentration results from rapid metabolism of VLDLs, which convert to LDLs as their triglyceride content is depleted through the action of lipoprotein lipase. However, advocates of the LMHR hypothesis have produced no evidence to date that LMHRs exhibit high rates of VLDL lipolysis or elevated lipoprotein lipase activity, both of which would have to be true for the model to hold. This VLDL-to-LDL conversion is absolutely essential for the lipid energy model to make sense, yet it has never been examined experimentally in this context, despite the availability of assays capable of doing so.
A simpler explanation for the rise in LDL-C in these patients is either or both of the following: 1) increased hepatic production and secretion of large, cholesterol-rich LDLs, or 2) decreased clearance of apoB, primarily LDLs, from plasma by the liver due to saturated fat induced suppression of the LDL receptor and increased cholesterol synthesis. However, the jugular question is not why apoB is elevated in these subjects, the critical question remains the same.
The critical question
The core issue in this debate is not whether LMHRs have unique metabolic characteristics, nor whether their elevated LDL-C arises from a different mechanism than other forms of hypercholesterolemia. The fundamental question is whether persistently elevated LDL-C and apoB levels contribute to atherosclerosis in these individuals, regardless of how or why those elevations occur.
As shown in the graph below (Figure 3), decades of research have consistently demonstrated that apoB-containing lipoproteins causally drive atherosclerosis. The mechanism is not abstract. It begins with arterial retention of apoB-containing particles, followed by their modification, uptake by macrophages, and propagation of a local inflammatory response that leads to plaque formation. Critically, each of these steps is a function of particle number and cumulative exposure over time. The arterial wall does not “interrogate” why apoB is elevated. It responds to how many particles enter and how long they remain. Any claim that a specific phenotype is exempt must therefore explain, at the level of arterial biology, why this sequence is interrupted. That explanation has not been provided.
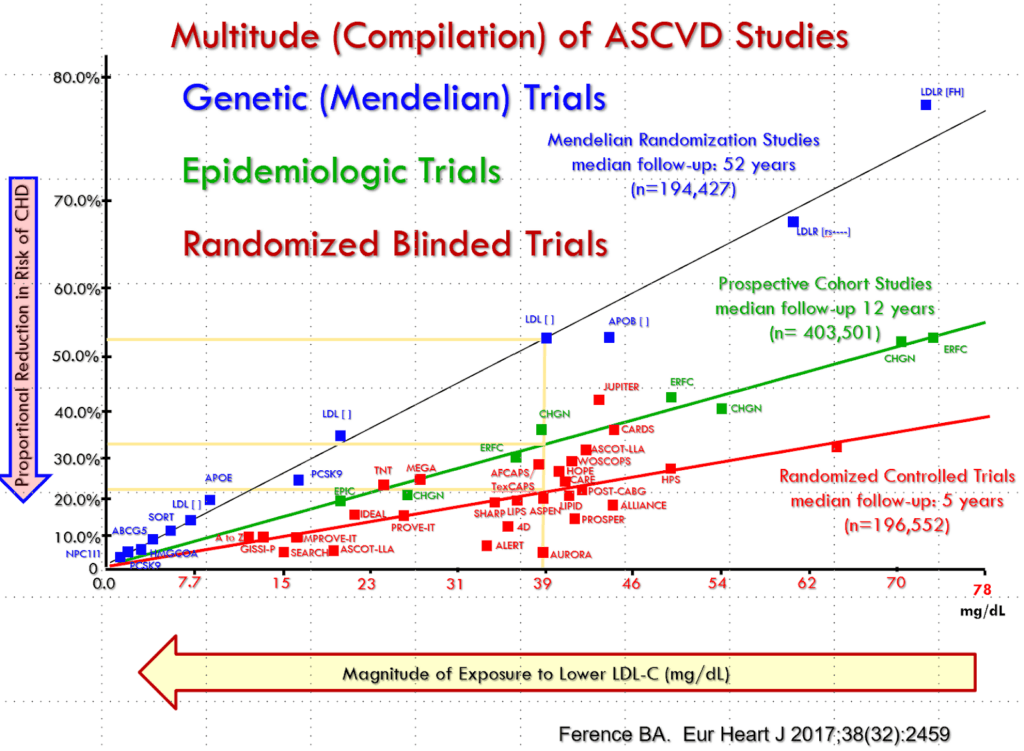
These conclusions are supported by (literally) hundreds of population studies, genetic studies such as Mendelian randomization, randomized controlled trials of LDL-lowering medications, and basic science research. Furthermore, if LDL is causal, it must be causal across all metabolic contexts unless a mechanism is shown that breaks that causality. For example, elevated apoB is atherogenic in familial hyper cholesterolemia (discussed in more detail below), whether the underlying mechanism is defects in LDL receptor function, changes in PCSK9 (which degrades those receptors), or because of defective apoB synthesis itself. Conversely, multiple interventions that lower apoB reduce ASCVD progression and events, whether the mechanism involves increasing LDL receptor expression by reducing cholesterol synthesis (statins, bempedoic acid), or inhibiting intestinal absorption of hepatobiliary excreted or dietary cholesterol (ezetimibe), decreasing hepatic LDL receptor turnover (PCSK9 inhibitors), or increasing hepatic LDL receptor activity (increasing the ratio of dietary polyunsaturated to saturated fatty acids).
In all cases, the effect on risk follows the effect on apoB concentration, independent of the mechanism whereby that delta is achieved. The burden of proof therefore lies with those claiming that LMHRs somehow escape this biology—that they are an exception to an extremely well-established rule. Two poorly designed observational cohorts are hardly sufficient to counter this mountain of evidence, especially when the competing mechanistic rationale is itself unsound.
In other words, the problem with the Keto CTA study runs deeper than its missing control group and short duration. Given that risk scales with cumulative apoB exposure, the absence of a short-term correlation in a relatively young, low-burden cohort is not evidence against causality. It is the expected result under a fully causal model.
In fact, the authors of the (now retracted and republished as a preprint) Keto CTA study admit this: In a response to concern about the Keto CTA study, they state “Moreover, our results are compatible with a causal role of apolipoprotein B in atherosclerosis, as we have openly acknowledged and supported in previous publications.”12
With that concession, the central dispute narrows considerably. The question is no longer whether apoB-containing lipoproteins are causal in atherosclerosis, but whether any specific metabolic context meaningfully attenuates that causal relationship. That is a much higher evidentiary bar. Demonstrating effect modification is fundamentally different from disputing causality itself, and it requires robust, long-term data showing that equivalent apoB exposure leads to different clinical outcomes. No such evidence currently exists. Absent that, acknowledging apoB as causal while implying that a subgroup is exempt is not a supported inference, it is an untested hypothesis.
Lessons from familial hypercholesterolemia
Proponents of the LMHR hypothesis often try to distinguish their population from individuals with familial hypercholesterolemia (FH), a group of genetic conditions that cause extremely high LDL-C levels and early development of ASCVD. Many FH patients have genetic defects in their LDL receptors, which are responsible for removing LDL from circulation via the liver. As a result, they clear LDL inefficiently, leading to markedly elevated levels of LDL-C and apoB. Advocates of the LMHR hypothesis argue that FH represents dysfunctional lipid metabolism, whereas LMHRs have “functional” elevated LDL-C, and they point to several distinctions between the two groups as evidence:
- LMHRs experience elevated LDL-C only on a ketogenic diet and have normal LDL-C on a mixed-macronutrient diet, whereas those with FH have elevated LDL-C regardless of diet composition.
- FH patients have elevated LDL regardless of BMI, whereas LDL levels in LMHRs are inversely correlated with BMI.
- HDL-C and triglyceride levels vary in FH, whereas in LMHRs HDL-C is consistently above 80 mg/dL and triglycerides are consistently below 70 mg/dL.
However, even if the high apoB in these subjects is the result of an adaptive response to fuel partitioning, that doesn’t make it “functional” in the sense of benign, any more than insulin resistance is “functional” because it’s the cell’s adaptive response to an overload of intracellular energy.
Some of these distinctions have additional flaws. For example, an inverse correlation between LDL-C and BMI in LMHRs does not account for the substantial variation in body composition possible at a given BMI, and therefore tells us little about a subject’s true metabolic state.
The distinction between “functional” and “dysfunctional” lipid metabolism is irrelevant. In familial hypercholesterolemia, apoB can be elevated through multiple mechanisms, including LDL receptor defects, PCSK9 variants, or altered apoB synthesis. In every case, risk tracks with apoB concentration, not with the mechanism producing it. The fundamental mechanism of atherosclerosis, the accumulation of apoB-containing lipoproteins in the arterial wall, does not depend on why the particles are elevated. If you’re facing a firing squad, it makes little difference whether the bullets hurtling toward your exposed torso are launched by single-shot, bolt-action, or semi-automatic rifles: You will always be better off with fewer of them.
The experience with FH patients provides some of our strongest evidence for the causal role of apoB and LDL-C in atherosclerosis. In addition to the differences listed above, advocates of the LMHR hypothesis point to one final distinction: While we know FH patients have high cardiovascular risk, we do not yet know the cardiovascular risk of LMHRs. But ironically, this “distinction” underscores the lack of evidence that high LDL-C is innocuous in individuals with an LMHR phenotype. FH has been studied over long periods, and FH patients have been exposed to high LDL-C and apoB since birth. In all cases, ASCVD is slow to develop. Heterozygous FH patients typically do not experience major adverse cardiac events until their third or fourth decade, and even children with homozygous FH and extremely elevated apoB usually do not suffer, with rare exceptions, adverse events for a decade or more after birth, meaning that it takes a long time to evaluate the risk conferred by apoB elevation. For years, many of these patients would also have normal CTAs. Thus, while proponents of the LMHR hypothesis like to draw distinctions between FH patients and LMHRs, they simultaneously reveal perhaps the greatest flaw in the LMHR concept: We simply have not had enough time to see the full long-term consequences of the LMHR profile.
Practical implications
Let us assume, for the sake of argument, that the LMHR phenotype accurately describes a small subset of the population. While prevention of ASCVD in the general population would typically call for an LDL-C level below 100 mg/dL, LMHRs often exhibit LDL-C levels two to six times higher. Yet even if an individual appears to fit the profile, we have no way of knowing with confidence whether that person is a “true” LMHR, nor do we have meaningful evidence that the long-term consequences of elevated LDL-C are any different in this population from any other population. Evidence from traditional observational studies, Mendelian randomization, and randomized clinical trials strongly and consistently indicates that apoB-containing lipoproteins drive atherogenesis, and that reducing apoB, or its liquid surrogate LDL-C, reduces the risk of atherosclerosis progression and of major adverse cardiovascular events such as heart attacks and strokes.
In response to the enormous volume of data linking high LDL-C with ASCVD, the CSF argues that data from the general population cannot be used to infer risk in LMHRs because the LMHR profile is rare in large population databases. Although very high apoB in general is not rare, affecting an estimated 1 in 200 people, the CSF argues that even among those with such hyperbetalipoproteinemia, those fitting the full LMHR phenotype are uncommon. But if this profile is truly so rare, is it really wise to assume that absence of evidence specific to this subpopulation is evidence of absence of harm? Excess concentration of apoB-containing lipoproteins is almost always atherogenic. As such, do we need specific evidence that elevated apoB is atherogenic in left-handed redheads with white blood cell counts below the reference range, Hungarian professional basketball players, and chess masters with an ALMI above the 90th percentile?
Ultimately, clinicians and patients are left with a practical question: What should we do when someone develops very high LDL-C on a low-carb, high-fat diet?
In the Cholesterol Code documentary, the principals suggest that people warning about the effects of high apoB on a ketogenic diet want to “take away” the choice to eat this way. That is a false dichotomy, and not our intent. One of us (Peter) spent three straight years on a ketogenic diet and got a lot of benefit from it, and we’ve racked up years of content on the benefits of ketogenic diets for the right patient, including our recent feature podcast on diets and our most recent interview with Dom D’Agostino.
Certainly, one option is to stop the ketogenic diet. But another is to remain on the diet, especially if it confers benefits such as weight management and improved metabolic health, but use lipid-lowering medications to return LDL-C to a safe range.
A third option, implied by some supporters of the LMHR hypothesis, is to continue on one’s merry way with sky-high LDL-C because meeting LMHR criteria might mean protection from the well-established atherogenic effects of elevated LDL. This option may seem the most appealing to many falling in the LMHR phenotype. Indeed, they likely feel great, do not have an active ASCVD diagnosis, and many other markers are favorable. In that last scenario, however, the only way to learn whether the assumption was wrong is to wait until it is too late and ASCVD has already taken hold. Indeed, toward the end of the documentary, one member of the clinical team responds to a question about this existential situation by suggesting that a person decide whether to lower their lipids based on the results of imaging of their arteries—that is, to only take action to prevent damage to your arteries once your arteries are already damaged. This is an unforced error, whether you’re on a ketogenic diet or not, as we discussed in a previous newsletter.
The decision framework here is asymmetric. The potential benefit of avoiding treatment is modest, while the downside risk of being wrong unfolds over years and can be irreversible. Atherosclerosis is cumulative and often clinically silent until late stages. By the time it is detectable by imaging or symptoms, the biological process is already well underway. In contrast, the tools available to lower apoB are numerous, well-characterized, and generally well tolerated. When evaluated through a risk management lens, the expected value calculation favors reducing apoB exposure rather than assuming, without evidence, that a specific phenotype confers protection. Even in the context of an otherwise healthy metabolic profile, the risks of dismissing elevated LDL-C as benign are simply too high, and the evidence for LDL-C’s role in atherosclerosis too strong, to justify relying on the LMHR hypothesis without extraordinary proof, which has not been provided.
With that said, does this mean metabolic health doesn’t matter? Of course not. Being metabolically healthy lowers your risk of many diseases, including ASCVD. We would not treat the opposite as true either (e.g., taking statins to lower apoB and therefore the risk of ASCVD does not automatically negate the risk of having high levels of visceral fat or insulin resistance). But that is beside the point for the current discussion. There are now multiple studies demonstrating that LDL-C continues to be monotonically associated with ASCVD, even in the absence of diabetes or other conventional cardiovascular risk factors.13–15 In fact, although unpublished, Dr. Deirdre Tobias of Tufts University has shown that higher LDL-C is associated with progressive elevations in all-cause mortality, even in people with low-risk features similar to LMHRs and more (HDL≥50, TG≤100, BMI<25, no hypertension, HbA1c<6.5, and nonsmoking).16 Metabolic health isimportant, but regardless of one’s metabolic health, it is not a free pass to ignore one of the primary drivers of CVD: apoB.
Looking forward
The LMHR hypothesis has stimulated valuable discussion about the relationship between diet, lipids, and cardiovascular risk. It may yet prove scientifically useful in helping us better understand this unusual lipid profile, the conditions under which it arises, and whether it differs in any meaningful way from more conventional forms of hypercholesterolemia. That evidence would require an empowered longer-term cardiovascular outcome trial which for all intents and purposes would be unethical. At most, it is plausible that favorable characteristics such as leanness, physical activity, and insulin sensitivity may mitigate some of the risk. While we should remain open to new evidence, the prudent approach is to continue taking elevated LDL-C seriously, regardless of context.
The central question is not whether LMHRs are metabolically unusual, nor whether their lipid profile arises by a route different from that of more conventional dyslipidemia. It is whether prolonged exposure to markedly elevated apoB-containing lipoproteins is benign in this population. At present, there is no compelling evidence that it is. Until such evidence exists, people trying to assess their own cardiovascular risk should follow the preponderance of the evidence, which still points in the same direction: Very high LDL-C is a problem to be addressed, not a gamble to be rationalized.
What would change my mind
I want to close by acknowledging something I think is essential to good scientific thinking.
My current position is a strong conviction: that apoB-containing lipoproteins are causally related to atherosclerosis, and that this relationship holds regardless of the context in which apoB is elevated. Put differently, my strong conviction remains that LDL, with its load of cholesterol, is causal under all circumstances with respect to atherosclerosis.
But strong convictions should be loosely held.
That means despite how strongly I speak on any subject, I am open to being wrong. It means I am open to new evidence. It means this belief can, in principle, be jostled out of my hands if the evidence warrants it. And I think that is how a good scientist should always think.
So what would it take to change my mind?
It would take clear, reproducible evidence that individuals with sustained, markedly elevated apoB, on the order of what we see in LMHRs, do not generate atherosclerosis over meaningful time horizons. Not one year. Not two years. Decades.
It would take well-designed prospective studies with appropriate control groups showing that LMHR individuals with apoB levels of 150, 180, or 200 mg/dL and above do not experience greater plaque progression or higher event rates than comparable individuals with lower apoB and otherwise similar baseline risk.
It would take mechanistic evidence showing that apoB-containing particles in this population behave differently at the level that matters most, the arterial wall.
In other words, for the LMHR hypothesis to be correct, one of two things must be true:
- ApoB-containing particles in these individuals do not enter or are not retained in the arterial wall, or
- They do enter but do not trigger inflammation and the multiple steps leading to plaque formation
There is currently no evidence for either claim, and both would require a fundamental revision of established atherosclerosis biology.
In addition, it would take a coherent, experimentally validated mechanism explaining why particle number does not translate into risk in this specific context.
And it would take all of that evidence to be consistent, reproducible, and durable over time.
You might think that that’s a very high evidential bar—and you’d be right. It’s the bar required for evidence to be strong enough to overturn decades of converging data from genetics, epidemiology, randomized trials, and basic science. Because what is at stake is not an abstract debate. It is whether people should feel comfortable ignoring one of the most well-established causal risk factors in medicine.
Until that level of evidence exists, my conviction remains. High apoB drives atherosclerosis. And very high LDL-C is not a curiosity to be rationalized. It is a risk to be taken seriously.

For a list of all previous weekly emails, click here.
References
1. Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem. 1994;45:303-369. doi:10.1016/s0065-3233(08)60643-9
2. Soffer DE, Marston NA, Maki KC, et al. Role of apolipoprotein B in the clinical management of cardiovascular risk in adults: An Expert Clinical Consensus from the National Lipid Association. J Clin Lipidol. 2024;18(5):e647-e663. doi:10.1016/j.jacl.2024.08.013
3. Blumenthal RS, Morris PB, Gaudino M, et al. 2026 ACC/AHA/AACVPR/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of Dyslipidemia: A report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. J Am Coll Cardiol. Published online March 13, 2026. doi:10.1016/j.jacc.2025.11.016
4. Norwitz NG, Cromwell WC. Oreo cookie treatment lowers LDL cholesterol more than high-intensity statin therapy in a Lean Mass Hyper-responder on a ketogenic diet: A curious crossover experiment. Metabolites. 2024;14(1):73. doi:10.3390/metabo14010073
5. Budoff M, Kinninger A, Manubolu V, Norwitz N, Feldman D, Soto-Mota A. The Impact of Sustained LDL-C Elevation on Plaque Changes: Primary Coronary plaque progression results from the Keto CTA Study. medRxiv. Published online January 16, 2026:2026.01.15.26343955. doi:10.64898/2026.01.15.26343955
6. Soto-Mota A, Flores-Jurado Y, Norwitz NG, et al. Increased low-density lipoprotein cholesterol on a low-carbohydrate diet in adults with normal but not high body weight: A meta-analysis. Am J Clin Nutr. 2024;119(3):740-747. doi:10.1016/j.ajcnut.2024.01.009
7. Norwitz NG, Soto-Mota A, Kaplan B, et al. The Lipid Energy Model: Reimagining lipoprotein function in the context of carbohydrate-restricted diets. Metabolites. 2022;12(5):460. doi:10.3390/metabo12050460
8. Javier DAR, Manubolu VS, Norwitz NG, et al. The impact of carbohydrate restriction-induced elevations in low-density lipoprotein cholesterol on progression of coronary atherosclerosis: the ketogenic diet trial study design. Coron Artery Dis. 2024;35(7):577-583. doi:10.1097/MCA.0000000000001395
9. Budoff M, Manubolu VS, Kinninger A, et al. Carbohydrate restriction-induced elevations in LDL-cholesterol and atherosclerosis: The KETO Trial. Metabolism. 2024;153(155854):155854. doi:10.1016/j.metabol.2024.155854
10. Garvey WT, Kwon S, Zheng D, et al. Effects of insulin resistance and type 2 diabetes on lipoprotein subclass particle size and concentration determined by nuclear magnetic resonance. Diabetes. 2003;52(2):453-462. doi:10.2337/diabetes.52.2.453
11. Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
12. Soto-Mota A, Norwitz NG, Manubolu VS, et al. Reply: The keto CTA study. JACC Adv. 2025;4(7):101862. doi:10.1016/j.jacadv.2025.101862
13. Masrouri S, Tamehri Zadeh SS, Shapiro MD, Khalili D, Hadaegh F. Impact of optimal cholesterol levels on subclinical atherosclerosis in the absence of risk factors in young adults. Atherosclerosis. 2024;393:117520. doi:10.1016/j.atherosclerosis.2024.117520
14. Faridi KF, Lahan S, Budoff MJ, et al. Serum lipoproteins are associated with coronary atherosclerosis in asymptomatic U.s. adults without traditional risk factors. JACC Adv. 2024;3(7):101049. doi:10.1016/j.jacadv.2024.101049
15. Fernández-Friera L, Fuster V, López-Melgar B, et al. Normal LDL-cholesterol levels are associated with subclinical atherosclerosis in the absence of risk factors. J Am Coll Cardiol. 2017;70(24):2979-2991. doi:10.1016/j.jacc.2017.10.024
16. [No title]. X (formerly Twitter). Accessed April 19, 2026. https://x.com/deirdre_tobias/status/1473636224258744322