Check out our 5-part series with Thomas Dayspring, M.D., FACP, FNLA, a world-renowned expert in lipidology:
- (October 15, 2018) Part I of V: An introduction to lipidology
- (October 16, 2018) Part II of V: Lipid metrics, lipid measurements, and cholesterol regulation
- (October 17, 2018) Part III of V: HDL, reverse cholesterol transport, CETP inhibitors, and apolipoproteins
- (October 18, 2018) Part IV of V: statins, ezetimibe, PCSK9 inhibitors, niacin, cholesterol and the brain
- (October 19, 2018) Part V of V: Lp(a), inflammation, oxLDL, remnants, and more
- (September 21, 2020) FOLLOW-UP: The latest insights into cardiovascular disease and lipidology
World-renowned lipidologist Tom Dayspring returns to give an update on the current thinking in lipidology as a follow-up to his 2018 five-part podcast series. In this episode, Tom discusses the growing consensus that atherogenic lipoproteins are essential drivers of atherosclerotic vascular disease. Tom further emphasizes apolipoprotein B (apoB) and lipoprotein(a) (Lp(a)). He provides insights into risk assessment, including which lab metrics to use, how to interpret them, and the appropriate therapeutic targets. Additionally, Tom discusses the most recent developments in lipid-lowering drug therapies—from the continued evolution of PCSK9 inhibitors, to the latest understanding of EPA and DHA, and the most recent addition of bempedoic acid to the list of therapeutic agents.
Subscribe on: APPLE PODCASTS | RSS | GOOGLE | OVERCAST | STITCHER
We discuss:
- The latest in the field of lipidology and cardiovascular disease [3:45];
- Apolipoproteins—the key to understanding lipid biology [9:30];
- ApoB as a preferred metric over LDL-P [16:30];
- Therapeutic goals for apoB concentration [21:45];
- Drivers of atherosclerosis [34:15];
- Overview and current thinking on high density lipoproteins (HDLs)—Is it a useful metric? [37:00];
- Lipoprotein(a)—the most dangerous particle you’ve never heard of [55:00];
- Are low density lipoprotein triglycerides (LDL-TGs) a useful metric? [1:13:15];
- Tom’s preferred lab measurements [1:17:45];
- The latest in lipid-lowering therapies [1:21:30];
- The different pathways among various lipid-lowering drugs [1:30:45];
- The latest on EPA and DHA [1:38:15];
- Fibrates—an underappreciated treatment for hypercholesterolemia [1:49:45] and;
- More.
Get Peter’s expertise in your inbox 100% free.
Sign up to receive An Introductory Guide to Longevity by Peter Attia, weekly longevity-focused articles, and new podcast announcements.The latest in the field of lipidology and cardiovascular disease [3:45]
Many patients are eager for in-depth information about lipidology
- The first podcast with Tom was the longest Peter has ever done (about 8 hours)
- There have exciting new developments in lipidology and CVD in the two years since the last podcast and we’ll go through those things today
Recent updates in lipidology
- Atherogenic lipoproteins are really the issue behind clinical atherosclerotic vascular disease
- The data have become overwhelming, so guidelines reflect this now
- Atherogenesis is sterol-mediated, but sterols are trafficked within apoB-containing lipoproteins, which is how they are transported into artery wall where they can start the pathological process
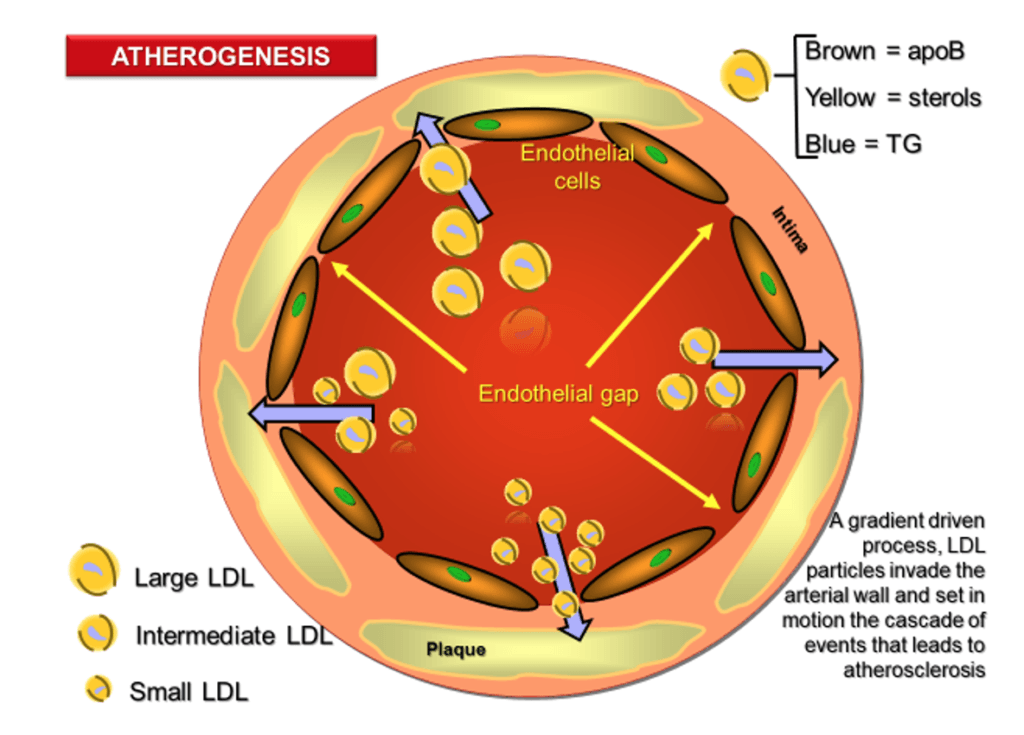
Figure 1. The process of atherogenesis.
- Atherogenic lipoproteins are still diagnosed using various cholesterol metrics, but ApoB is within every guideline now (Allan Sniderman has been advocating for this for a long time)
- What contributes to the atherogenicity of lipoproteins?
- TGs have taken center stage – affect concentration and functionality

Figure 2. Lipoproteins trafficking triglycerides.
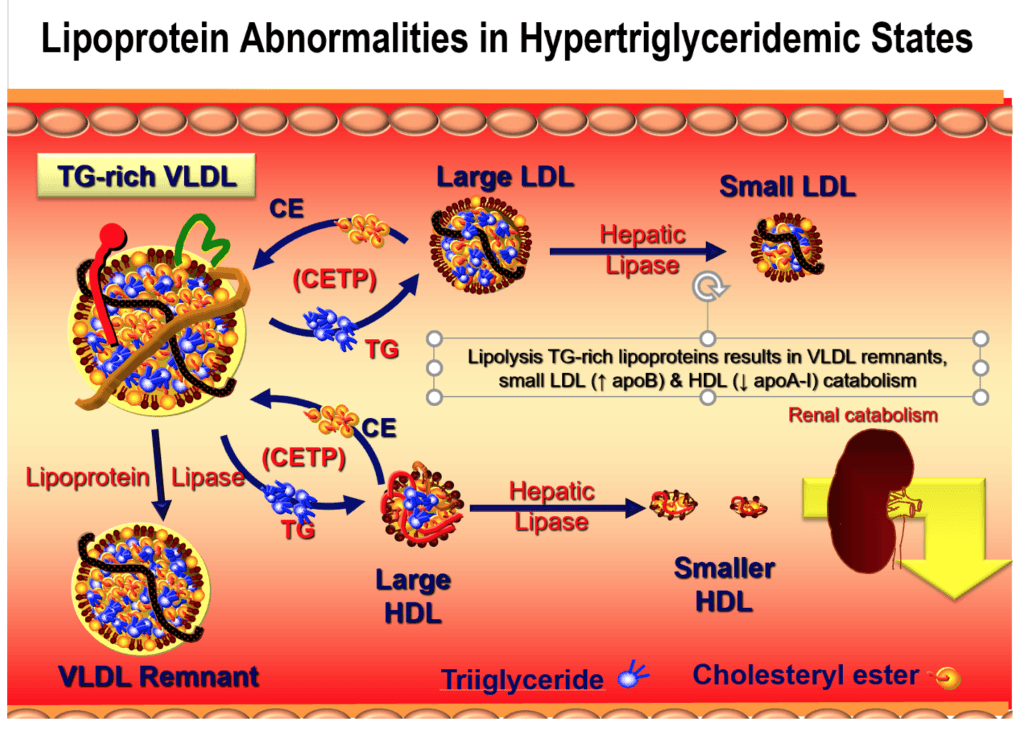
Figure 3. Lipoprotein structure with high TG levels.
- HDL is no longer considered informative to use
- Lp(a) has emerging significance
- Pharmacology has also advanced
***
Topics on today’s agenda:
- Atherogenic lipoproteins (apoB/LDL-P) are front and center in pathogenesis of CVD
- Methods of risk assessment (e.g., HDL no longer primary)
- What’s developed in therapies – more data around ezetimibe, PSCK9 inhibitors, omega 3 fatty acids, and a few other treatments (expanding on what we spoke about with Bill Harris)
Apolipoproteins—the key to understanding lipid biology [9:30]
ApoB & LDL-P are used interchangeably, but this is not quite accurate.
{end of show notes preview}
Would you like access to extensive show notes and references for this podcast (and more)?
Check out this post to see an example of what the substantial show notes look like. Become a member today to get access.

Thomas Dayspring, M.D., FACP, FNLA
Thomas Dayspring, MD, FACP, FNLA is the chief academic officer for True Health Diagnostics, LLC. He provides scientific leadership and direction for the company’s comprehensive educational programs. Dr. Dayspring is a fellow of both the American College of Physicians and the National Lipid Association. He is certified in internal medicine and clinical lipidology.
Before relocating to Virginia in 2012, Dr. Dayspring practiced medicine in New Jersey for 37 years. Over the last two decades, he has given over 4,000 domestic and international lectures, including over 600 CME programs on topics such as atherothrombosis, lipoprotein and vascular biology, biomarker testing, and women’s cardiovascular issues.
Dr Dayspring is an Associate Editor of the Journal of Clinical Lipidology. He has authored or co-authored numerous manuscripts published across leading journals such as the American Journal of Cardiology, the Journal of Clinical Lipidology, and several lipid-related book chapters. He was the recipient of the 2011 National Lipid Association President’s Award for services to clinical lipidology. [truehealthdiag.com]
Disclosures:
- Employed full time for last three years by True Health Diagnostics, LLC, which provides biomarker diagnostics and clinical services to clinicians, patients, and healthcare organizations
- 2017: small consulting project for Abbvie
Tom on Twitter: @Drlipid